Apple (Malus domestica) is one of the most economically important fruits, with more than 31,000 ha cultivation area in South Korea [1]. However, numerous destructive pathogens infect apple trees and significantly reduce the commercial apple production worldwide. Among the fungal diseases, canker, twig dieback, and plant decline frequently occurs in apple orchards [2]. During the apple cultivation period, apple decline symptoms were observed from May until the end of September 2020 in apple (cv. Fuji) orchards located in Gyeonggi, Gyeongbuk, and Gangwon provinces. Mostly, declined trees were young (less than ten years), showing warts on the surface, darkening from the trunk, detachment of epidermis of the stem, embedded pycnidia on the bark, withering of branch, and eventually leading to decline (Fig. 1A-1G, and I). In this present study, isolated fungal strains were described and illustrated as a causal agent of apple decline.

Fig. 1. Natural apple decline symptoms caused by Botryosphaeria sinensis and description of the KNUF-20-014. A, declined apple tree; B, warts on a diseased tree; C and D, blackened trunk and observed symptoms inside; E and F, black dots on a diseased trunk; G, observed necrosis on the internal of a diseased tree; H, pathogenicity test result shows canker and necrosis on the inoculated branch of cv. Fuji and its internal symptom (red arrows indicate inoculated zones); I and J, pycnidia on a diseased tree and on pine needle agar (PNA) medium; K, cross-sections through pycnidium; L and M, colony on malt extract agar after incubation at 28℃ for five days (L, front; M, reverse); N, conidia (scale bar=10 μm).
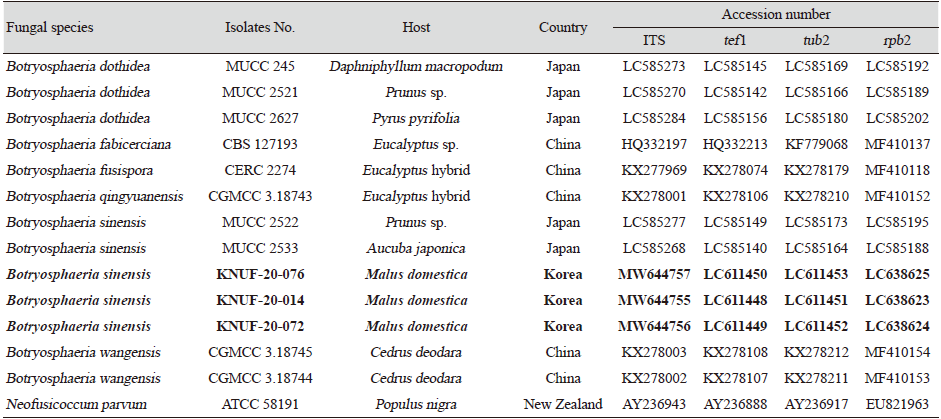
To isolate the causal fungus, the fragment (approximately 2 mm) was taken from the margin of internal lesions, transferred onto potato dextrose agar (PDA; Difco, Detroit, MI, USA), and incubated at 25℃. As a result, the three strains, KNUF-20-014, KNUF-20-072, and KNUF-20-074, were isolated from the diseased apple tree. For molecular identification of strains at the genus and species levels, the total genomic DNA was extracted from the isolates using a HiGene Genomic DNA prep kit (BIOFACT, Daejeon, Korea) according to the manufacturer’s instructions. Molecular identification of the strains was conducted using the nucleotide sequences of internal transcribed spacer (ITS) regions and translation elongation factor 1-alpha (tef1), beta-tubulin (tub2), and second largest subunit of RNA polymerase II (rpb2) genes. The ITS regions, tef1, tub2, and rpb2 genes were amplified using the primer sets ITS1/ITS4 [3], EF1-668F/EF1-1251R [4], Bt2a/Bt2b [5], and RPB2-5F2/fRPB2-7cR [6], respectively. Amplified products of the polymerase chain reaction (PCR) were purified with EXOSAP-IT (Thermo Fisher Scientific, Waltham, MA, USA) and sequenced by Macrogen Co. Ltd. (Daejeon, Korea). Amplification of the ITS, tef1, tub2, and rpb2 loci of strains KNUF-20-014, KNUF-20-072, and KNUF-20-074 yielded fragments of 526, 512, 413, and 1,020 bp; 520, 500, 422, and 1,005 bp; and 497, 496, 423, and 1,029 bp, respectively. The comparative sequence analyses of the molecular markers of the three strains revealed their similarity of 100%, indicating their affiliation to the same species. A BLAST search of the NCBI database showed that the sequences obtained from ITS, tef1, tub2, and rpb2 loci of the three strains exhibited highest similarities of 99.8% (517 bp out of 518 bp), 100% (267 bp out of 267 bp), 100% (363 bp out of 363 bp), and 100% (577 bp out of 577) with that of Botryosphaeria sinensis MUCC 2533 (accession numbers LC585268, LC585140, LC585164, LC585188, respectively). To confirm the closest relationship between the three strains and B. sinensis at the species level, a phylogenetic analysis was conducted using concatenated sequences of the ITS regions, tef1, tub2, and rpb2 genes. The sequences of allied species were retrieved from the NCBI database (Table 1). A phylogenetic tree was constructed with maximum-likelihood method using the program MEGA7 [7]. The evolutionary distances were calculated using the Kimura two-parameter model [8] and the topology of the tree was evaluated using bootstrap analysis based on 1,000 replicates. In the phylogenetic tree, the isolated strains occupied a position within the genus Botryosphaeria and clustered together with B. sinensis, indicating their closest relationship at the species level (Fig. 2).
The representative isolate, KNUF-20-014, was cultured on pine needle agar (PNA) and malt extract agar (MEA) to check the cultural and morphological characteristics. On MEA the colonies grew up to 80 mm in three days at 28℃, showing gray aerial mycelium, white and gray surface with an olivaceous black reverse (Fig. 1L and 1M), the pycnidia were produced on PNA after 21 days (Fig. 1J and 1K), conidia were hyaline, aseptate, obtuse apex, fusiform, smooth with granular contents, and 19.7-28.9×4.9-7.3 μm (av. 23.4×6.0 μm, l/w 4.1, n=50) (Fig. 1N). These cultural and morphological characteristics were similar to those of the previously described B. sinensis [9], but strain KNUF-20-014 was readily distinguishable from B. dothidea by its conidia size. The average conidia length of the strain (23.4 μm) was distinctly shorter than that of B. dothidea (26.2 μm), while the width of its conidia (6.0 μm) was longer than that of B. dothidea (5.4 μm) [10]. The conidial length:width ratio of strain KNUF-20-014 (4.1) was the same as the reported value for B. sinensis [9], but clearly different from that of B. dothidea (4.9) [10].

Fig. 2. Maximum-likelihood phylogenetic tree, based on internal transcribed spacer (ITS) regions, translation elongation factor-1 (tef1) , beta-tubulin (tub 2) , and RNA polymerase II (rpb 2) gene sequences, showing the phylogenetic position of strains KNUF-20-076, KNUF-20-014, and KNUF-20-072, among related strains of the genus Botryosphaeria . Bootstrap values greater than 60% (percentage of 1,000 replications) are shown at branching points. The tree was rooted using Neofusicoccum parvum ATCC 58191 as an outgroup. Bar, 0.01 substitutions per nucleotide position.
Currently, there are controversies regarding the classification of a few species of the genus Botryosphaeria [11,12]. Based on phylogenetic analysis using the three combined ITS, tef1, and tub2 sequences, B. sinensis [9], B. minutispermatia [13], B. quercus [14], B. qinlingensis [15], and B. wangensis [16] were reclassified as a later synonym of B. dothidea by Zhang et al. [12]. At the same time, Hattori et al. [11] reexamined the genus Botryosphaeria based on phylogenetic analyses using the four molecular markers, namely ITS, tef1, tub2, and rpb2, and showed that B. sinensis and B. dothidea represent separate species. Simultaneously, the conidia size was highlighted as a key morphological characteristic for the differentiating species in the genus Botryosphaeria. Typically, an increase in the number of genes used in multi locus sequence analysis (MLSA) resulted in more accurate identification of isolated strains, therefore, we conducted MLSA using the four molecular markers in our study. According to the phylogenetic analysis, cultural and morphological characteristics, KNUF-20-014 was identified to be the same with B. sinensis at the species level but differed from B. dothidea. These data are in good agreement with the results of the reexamination of the genus Botryosphaeria conducted by Hattori et al. [11].
A pathogenicity test of strain KNUF-20-014 was conducted with healthy branches cv. Fuji. The branches were inoculated by placing a mycelial plug (4-5 mm) from the five-day-old colony on fresh wound sites made with a sterilized needle, while PDA plugs were placed into similar wounds on the three branches as mock inoculation. The inoculation points were wrapped in parafilm to maintain the moisture for three days at 25℃ in a growth chamber. After four weeks, all inoculated branches showed dark-brown discolorations and vascular necrosis, whereas no symptoms were observed in the mock inoculated branches (Fig. 1H). Pathogenicity test was conducted three times, and the pathogen was re-isolated from the inoculated branches.
Botryosphaeria species are known as plant saprobes, pathogens, and endophytes, with global distribution, on a wide variety of mainly woody hosts [13,17]. Diseases caused by pathogens belonging to the genus Botryosphaeria have resulted in significant losses in various economically important agricultural crops. Apple black rot caused by B. obtusa has resulted in fruit loss of 25-50% in the USA [18] and 20% annual losses of vineyard productivity from B. stevensii infection were reported in France [19]. Among them, B. dothidea is known to be a serious pathogen mainly of woody plants that were reported in 66 countries and confirmed in more than 24 host genera [20]. In Korea, B. dothidea has been previously reported to cause canker and warts on infected apple branches, mainly occurring on the cv. Hongro and rarely observed on the cv. Fuji [21,22]. However, in this study, B. sinensis was mainly isolated from the cv. Fuji, which could be related to host specificity.
B. sinensis was recorded on the twigs of Populus sp., Morus alba, Juglans regia in China [9], Paulownia tomentosa and Prunus sp. in Japan [11], and recently identified on Mangifera indica in Australia [23]. Our results increase the awareness of Botryosphaeria distribution, thereby improving our understanding of apple decline associated with B. sinensis and can be used for developing the control methods to prevent economic losses.

