INTRODUCTION
Macrofungi are a group of filamentous fungi that produce distinctive spore-bearing structures called fruiting bodies. In ecosystems, they play essential roles as decomposers and recyclers [1,2]. They are largely consumed as foods by humans due to their nutritional value and unique flavors [3,4]. In addition, accumulating evidence has shown that macrofungi possess numerous bioactivities such as antioxidative, anti-inflammatory, antidiabetic, and anticancer activities [3,5,6]. Elaborate research has been performed to elucidate the responsible compounds and their chemical structures, further supporting the medicinal potential of macrofungi [7-13]. Meanwhile, bioremediation capability of macrofungi is attracting increasing attention. In particular, non-specific ligninolytic enzymes secreted by white-rot basidiomycetes are reported to be capable of degrading toxic organic pollutants such as polycyclic aromatic hydrocarbons (PAHs) and azo-type and anthraquinone-type textile dyes [3,14]. It has also been documented that plastics, such as low-density polyethylene (LDPE), could be biodegraded by Pleurotus ostreatus [15,16].
To exploit the above-mentioned potential of macrofungi, it is necessary to explore the valuable activities of diverse macrofungi. A proper identification of macrofungi is the first step in this process. In addition, studies to reveal the specific gene function followed by genetic modification and engineering are also needed to enhance the effectiveness of macrofungi. Amplification of specific DNA fragments is fundamental to both genetic approaches and fungal identification [17,18].
Conventional genomic DNA extraction methods, such as sodium dodecyl sulfate (SDS) and cetyltrimethylammonium bromide (CTAB) methods, provide reliable template DNA. However, these methods are laborious and time-consuming, especially when handling a large number of samples. They also require hazardous materials such as phenol and chloroform. Commercial DNA isolation kits partially overcome this problem by simplifying the procedure and using a solid-phase column instead of organic solvents; however, they are still time-consuming and expensive. In addition, both conventional and commercial kit-based methods require relatively large amounts of raw materials.
Direct PCR amplifies DNA directly from the raw materials. Because the direct PCR method omits the laborious process of DNA isolation, it reduces time and costs, and is therefore suitable for high-throughput approaches. Moreover, due to the requirement of low sample amount, it alleviates the risk of DNA isolation failure and loss of sample. It has been developed for many organisms, including bacteria [19], algae [20], plants [21,22], and fungi [23,24]. Although there are several reports on direct PCR for macrofungal species [25-27], these studies were performed with a limited number of species. For instance, Bonito presented a direct PCR method for five different truffle species [25]. Izumitsu et al. developed a method using microwaving and adopted the method for eight mushroom species [26]. Wang et al. reported a method applicable to three macrofungal species for transformant screening [27].
In this study, we established a rapid and efficient direct PCR method for macrofungi and showed that this method is widely applicable to various macrofungal species (63 different species), including both ascomycetes and basidiomycetes. Using this method, we successfully amplified not only high-copy internal transcribed spacer (ITS) fragments, but also low-copy and large-sized genes. This method is expected to be applied to molecular identification, DNA marker typing, gene manipulation, and transformant screening of macrofungi.
MATERIALS AND METHODS
Strains and culture conditions
All fungal isolates used in this study were obtained from the National Institute of Forest Science (NIFoS), including the monokaryotic strain derived from basidiospore of Sanmaru 1ho (NIFoS 2924), a cultivar strain of L. edodes. A total of 14 species of ascomycetes and 49 species of basidiomycetes were included in the assays. The obtained isolates were grown on potato dextrose agar (PDA; BD Difco, MD, USA) medium at 25℃ under dark conditions for 2 to 3 weeks.
Lysate preparation
A small amount of mycelia (approximately 3 mm) was scratched using a sterilized micropipette tip. For the TE method, the mycelia were resuspended in 50 μL of TE (10 mM Tris and 1 mM EDTA, pH 8.0) and heated with occasional vortexing. For the NEU method, the mycelia were resuspended in 25 μL of 20 mM KOH and incubated at high temperatures with occasional vortexing. Then, 25 μL of 10 mM (NH4)2SO4 was added for neutralization. For the TE/NEU method, the mycelia were resuspended in a mixture of 40 μL of TE and 5 μL of 200 mM KOH and heated with occasional vortexing. Then, 5 μL of 100 mM (NH4)2SO4 was added for neutralization. To examine the effects of the lysis methods, mycelia resuspended in different solutions were heated at 65℃ for 10 min. To investigate the effects of lysis temperature, mycelia were resuspended in a mixture of 40 μL of TE and 5 μL of 200 mM KOH, and were heated at 50, 65, 80, or 95℃ for 10 min; (NH4)2SO4 was then added for neutralization. To examine the effects of heating time, mycelia resuspended in a mixture of 40 μL of TE and 5 μL of 200 mM KOH were heated at 65℃ for 5, 10, 20, or 30 min, and then (NH4)2SO4 was added for neutralization. The resultant lysates were centrifuged at 13,000 × g for 1 min, and the supernatants were used for PCR amplification.
DW without DNA was used as the negative control. For the positive control, genomic DNA was extracted using DNeasy Plant Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions.
PCR analysis
For conventional PCR analysis, 2 μL of lysate was used for PCR amplification in a reaction volume of 20 μL containing 1 μL of 10 μM of each primer, 2 μL of 10X reaction buffer (100 mM Tris, 400 mM KCl, and 15 mM MgCl2, pH 9), 1 μL of 10 mM dNTP, and 1 unit of Taq DNA polymerase (Bioneer, Daejeon, Korea). PCR reactions were performed with a C1000 thermal cycler (Bio-Rad, CA, USA) under the following conditions: 95℃ for 5 min, 23-35 cycles of 95℃ for 30 s, 55℃ for 30 s, 72℃ for 30–120 s, and 72℃ for 10 min. Five microliters of each PCR product was examined by agarose gel electrophoresis.
For real-time PCR analysis, 2 μL of lysate was used for PCR amplification in a reaction volume of 20 μL containing 1 μL of 10 μM of each primer and 2X SsoAdvanced Universal SYBR Green Supermix (Bio-Rad). PCR was conducted using a CFX Connect Real-Time System (Bio-Rad) under the following conditions: 95℃ for 3 min; 40 cycles of 95℃ for 10 s, and 60℃ for 30 s. Relative quantity was calculated using the delta Ct method. To statistically compare the relative quantities, one-way ANOVA followed by Duncan’s multiple-range test (p<0.05) was performed using SPSS software (SPSS version18, SPSS IBM, NY, USA).
To investigate the effects of PCR additives, betaine (Sigma-Aldrich, MO, USA), bovine serum albumin (BSA; Sigma-Aldrich), dimethyl sulfoxide (DMSO; Sigma-Aldrich), gelatin (Sigma-Aldrich), glycerol (Junsei Chemical, Tokyo, Japan), and Tween-20 (Yakuri Pure Chemicals, Kyoto, Japan) were added to the reaction mixtures. Except betaine, stock solutions of PCR additives were prepared at 10×concentration, and 2 μL of stock was used for PCR amplification in a reaction volume of 20 μL. Because of the low solubility of betaine, a 5 M stock solution was prepared; 4 or 8 μL of stock was used for PCR amplification in a reaction volume of 20 μL for attaining 1 or 2 M final concentration, respectively. The primers used for PCR amplification are listed in Fig. S1.
Sequence analysis
The PCR products were purified from agarose gel using AccuPrep PCR/Gel DNA Purification kit (Bioneer) and then cloned into a TA vector using the TOPcloner TA kit (Enzynomics, Daejeon, Korea) following the manufacturer’s instructions. Sequencing of the clones was conducted by Macrogen (Seoul, Korea) using the Sanger method with an ABI 3730xl DNA Analyzer (Applied Biosystems, CA, USA). The determined sequences were analyzed using the NCBI nucleotide BLAST with default parameters (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
RESULTS
Optimization of lysis method
To establish an efficient lysis method, we first examined the effects of lysis solutions, including negative and positive controls (Fig. 1A). PCR with the lysate extracted using distilled water (DW) failed to amplify ITS fragments under our assay conditions. PCR with lysates extracted using TE buffer and KOH as an alkali solution produced amplification of ITS fragments with occasional failure. The TE/NEU method using both TE buffer and KOH showed the most successful amplification of the ITS fragments. To quantitatively compare the effects of lysis solutions, we performed real-time PCR using primer sets specific to the ITS region of L. edodes. PCR with lysates prepared using TE, NEU, and TE/NEU methods resulted in markedly higher amplification than that with lysates prepared using DW (Fig. 1B). The TE/NEU method showed a statistically significant increase in the amplification as compared to the TE method. There was no statistically significant difference between the amplification obtained using TE/NEU and NEU methods. Considering the stability of DNA in TE buffer, we decided to adopt the TE/NEU method for further experiments.
Next, we investigated the effects of the lysis temperature and heat treatment time. In both conventional and real-time PCR conditions, incubation at 65℃ produced the highest amplification (Fig. 1C and 1D). Therefore, we subsequently examined the effect of different heat treatment times at 65℃. Under conventional PCR conditions, we found that 5 to 20 min treatment was effective for the amplification of ITS fragments and a 30 min treatment resulted in relatively poor amplification (Fig. 1E). Under real-time PCR conditions, we did not observe any statistically significant differences among the tested treatment times, but the 10 min treatment showed the highest average value (Fig. 1F).
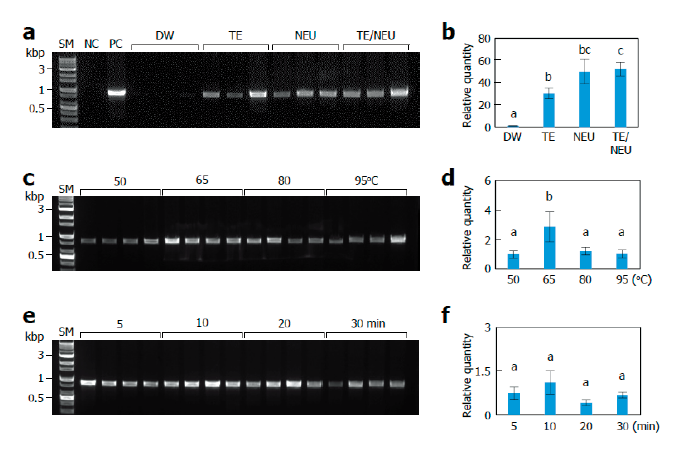
Fig. 1. Effects of lysis conditions. Monokaryotic mycelia of Sanmaru 1ho were lysed with different methods. The resultant lysates were subjected to internal transcribed spacer (ITS) region amplification with conventional PCR (A, C, D) or real-time PCR (B, D, E). The relative quantities of DW, 50℃, and 5 min were set to 1 as reference. The letters, a-c, indicate statistically significant differences based on one-way ANOVA followed by Duncan’s multiple-range test (p<0.05). Bars, S.E. (A, B) Effects of lysis solutions. SM, size marker; NC, negative control; PC, positive control; DW, distilled water; TE, Tris/EDTA; NEU, neutralization of KOH with (NH4)2SO4; TE/NEU, neutralization of Tris/EDTA/KOH with (NH4)2SO4. (C, D) Effects of lysis temperatures. (E, F) Effects of heat treatment times.
Improvement in PCR amplification using additives
To increase the efficiency of the direct PCR method, we decided to use various chemicals known as PCR additives [28-33] and evaluate their effects. In conventional PCR conditions, bovine serum albumin (BSA), gelatin, and dimethyl sulfoxide (DMSO) were found to be effective in enhancing the amplification of ITS fragments at some concentrations, whereas glycerol, betaine, and Tween-20 showed negative or negligible effects on the ITS amplification at the tested concentrations (Fig. 2A and 2C). However, we could not find any statistically significant effects of the PCR additives, except for the negative effects of betaine and Tween-20 at high concentrations in real-time PCR conditions (Fig. 2B and 2D). Different effects of PCR additives in conventional and real-time PCR conditions may arise from the difference in DNA polymerases used (see Discussion).
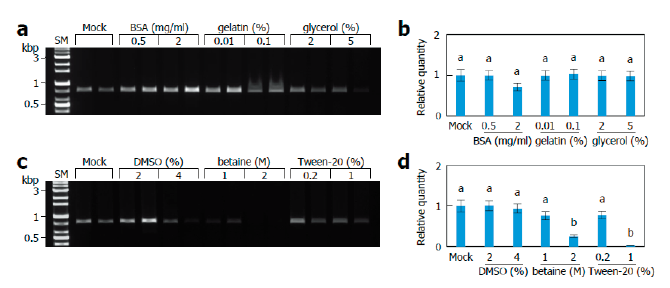
Fig. 2. Effects of PCR additives. Monokaryotic mycelia of Sanmaru 1ho were lysed with TE/NEU method and then the resultant lysates were subjected to internal transcribed spacer (ITS) region amplification with conventional PCR (A, C) or real-time PCR (B, D) in the presence of different PCR additives. The relative quantity of Mock was set to 1 as reference. The letters, a and b, indicate statistically significant differences based on one-way ANOVA followed by Duncan’s multiple-range test (p<0.05). Bars, S.E. SM, size marker; BSA, bovine serum albuminD; MSO, dimethyl sulfoxide.
Application to low copy gene amplification
Next, we performed direct PCR to amplify the pheromone receptor (rcb) genes of L. edodes to evaluate whether this method is applicable for the amplification of low copy genes as well as multi-copy genes [34]. Because the addition of BSA markedly increased the amplification of ITS fragments under conventional PCR conditions, we decided to conduct the direct PCR by sequentially performing TE/NEU lysis and BSA-added PCR amplification for further experiments. Using direct PCR method, we successfully identified the B5 mating type of the monokaryotic strain of Sanmaru 1ho because it contained rcb1-2 and rcb2-2 alleles (Fig. 3A).
We also applied this method to amplify low-copy and large-sized genes. Approximately 3 kb long genes, namely mitochondrial intermediate peptidase (MIP), LACCASE 4 (LCC4), and PHOTORECEPTOR A (PHRA), were successfully amplified (Fig. 3B), and the specificity of amplicons was verified by sequencing and alignment with the reported sequences (Fig. S2, S3, and S4) [35-37]. By comparing the genomic sequence obtained by direct PCR and cDNA sequence reported previously, we were able to predict the intron structures of the LCC4 gene of Sanmaru 1ho (Fig. S3) [36]. We also identified mutations in the PHRA gene of Sanmaru 1ho, with insertion of 2 amino acids (Fig. S4) [37]. Taken together, direct PCR could amplify low-copy genes as well as multi-copy genes, and therefore, it may be employed for DNA marker typing and gene cloning.
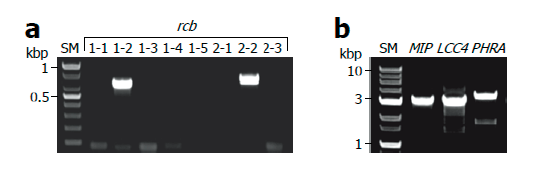
Fig. 3. Amplification of low copy genes. Monokaryotic mycelia of Sanmaru 1ho were lysed with TE/NEU method and then the resultant lysates were subjected to PCR amplification in the presence of bovine serum albumin (BSA) at final concentration of 2 mg/ml. (A) Amplification of pheromone receptor (rcb) genes in B mating type locus. (B) Amplification of low copy and large-sized genes. SM, size marker; MIP, mitochondrial intermediate peptidase; LCC4, LACCASE 4; PHRA, PHOTORECEPTOR A.
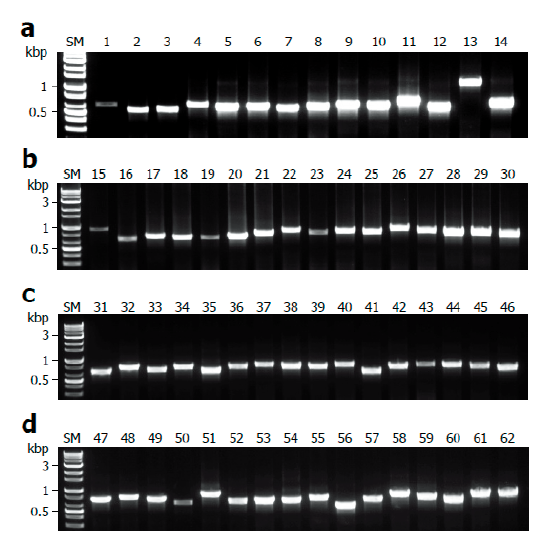
Fig. 4. Application for a variety of macrofungi. Mycelia of ascomycetes and basidiomycetes were lysed with TE/NEU method and then the resultant lysates were subjected to internal transcribed spacer (ITS) region amplification in the presence of bovine serum albumin (BAS) at final concentration of 2 mg/ml. (A) Amplification of ITS regions of ascomycetes. (B-D) Amplification of ITS regions of basidiomycetes. SM, size marker.
Application to various macrofungal species
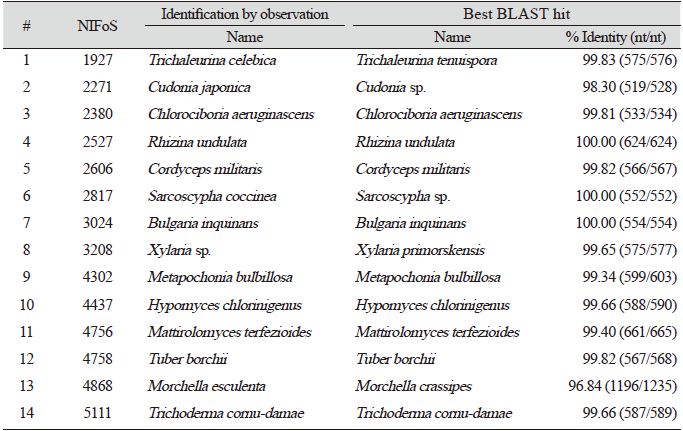
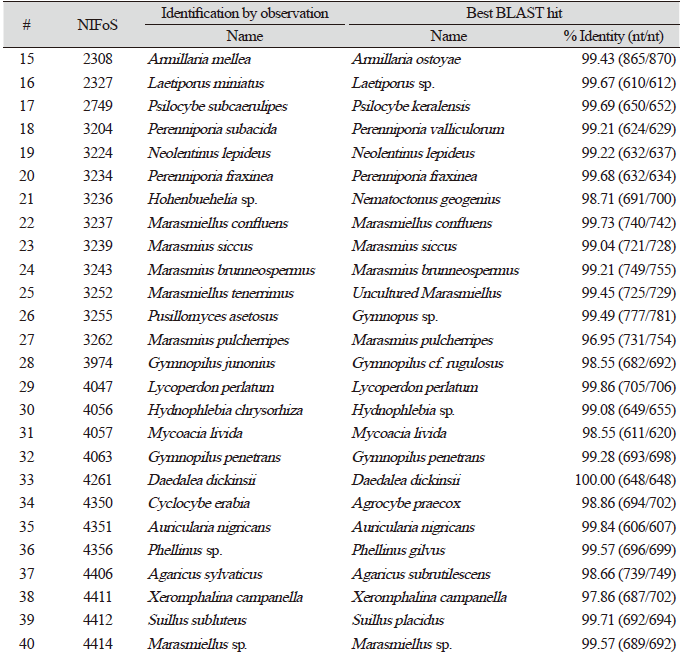
Finally, we evaluated the universality of this method for various macrofungi, including 14 species of ascomycetes and 48 species of basidiomycetes. We incorporated commercially important mushroom species, such as Cordyceps militaris (lane 5) and Pleurotus ostreatus (lane 49) in the assays, and they showed a high level of amplification (Fig. 4). Although there were some variations in the levels of amplification (Fig. 4), 62 different species and 54 different genera of macrofungi were successfully identified based on the sequences of amplified ITS fragments using this method (Tables 1 and 2). These results indicate the wide-range applicability of this method.

DISCUSSION
In this study, we established a direct PCR method that bypassed the laborious and time-consuming DNA isolation process and demonstrated its applicability in various macrofungal species. This direct PCR method has several advantages over conventional DNA isolation methods, in addition to time- and cost-saving properties. Because of its simple procedure, it minimizes the possibility of human error and contamination. It also reduces the risk of amplification failure and loss of sample because only a small amount of sample material is required at once. The requirement of low sample amount allows the identification of species or transformants at the early stage of mycelial growth and therefore decreases the burden of culturing unintended cells. Moreover, it can preserve precious sample materials of slowly growing or hard-to-culture species such as ectomycorrhizal macrofungi.
We used several chemicals and PCR additives in this method. First, a mixture of KOH and TE buffer was used for cell lysis. Alkali treatment can solubilize the polysaccharides present in the cell walls of both ascomycetes and basidiomycetes [38,39]. EDTA is known to inhibit DNase activity by chelating divalent cations. The combinatorial use of a strong base for cell wall lysis and EDTA for DNase inhibition seems to promote the successful release of DNA. Second, we used (NH4)2SO4 as the acid for neutralization. Because the lysate is directly included in PCR reaction mixtures, neutralization after alkaline lysis should be performed to adjust the pH for optimum DNA polymerase activity. Ammonium ion (NH4+) has been reported to improve the specificity and efficiency of PCR by destabilizing non-specific interactions between the primer and template DNA [40,41]. Dual functions of (NH4)2SO4 as an acid and a PCR enhancer may contribute to the successful amplification. Neutralization using other acids, such as HCl, can further clarify the function of (NH4)2SO4 as a PCR enhancer in this method. Third, BSA was included in the PCR reaction mixture. We examined the effects of several chemicals that are known to enhance DNA amplification by multiple modes of action. For instance, BSA and gelatin have been reported to stabilize the DNA polymerase and decrease the inhibitory effects of secondary metabolites [28-30]. Glycerol, DMSO, and betaine have been reported to enhance the amplification by reducing the secondary structure of template DNA containing GC-rich regions [30-32]. Tween-20, a non-ionic detergent, stabilizes DNA polymerase and hinders the secondary structure formation of template DNA [33]. Among the PCR additives examined, BSA addition exhibited the most reliable and increased amplification results in Taq polymerase-based conventional PCR conditions (Fig. 2). BSA is known to be beneficial for overcoming multiple PCR inhibitors and stabilizing Taq DNA polymerase [28,30]. Therefore, BSA might promote PCR amplification by repressing the inhibitory effects of various metabolites in the crude lysate of macrofungi.
Although we investigated the effects of a single PCR additive, there are reports on the combined use of multiple PCR enhancers [42-44]. Considering the diversity of metabolites across the macrofungal species, combinatorial use of PCR additives could further enhance the efficiency of direct PCR for macrofungi.
While the addition of BSA showed positive effects in Taq polymerase-based conventional PCR conditions, it displayed no significant effects under our real-time PCR conditions (Fig. 2). One possible explanation for this discrepancy is the difference in DNA polymerases. In our real-time PCR assay, DNA polymerase fused with Sso7d (a small DNA-binding protein) was used. Fusion of Sso7d to DNA polymerase has been reported to enhance the processivity of polymerase, thereby increasing the efficiency of PCR [21,45]. It seems that the effects of Sso7d fused with DNA polymerase overwhelmed those of BSA addition on PCR efficiency under our assay conditions.
Meanwhile, we found that the effects of lysis solutions were consistent in Taq polymerase-based conventional PCR conditions and Sso7d fused polymerase-based real-time PCR conditions (Fig. 1). These data indicate that different lysis solutions result in different amounts of DNA release. Moreover, the data support that our lysis method is applicable to both conventional and real-time PCR.
CONCLUSION
In conclusion, we established a rapid and efficient method of direct PCR for a variety of macrofungi, including both ascomycetes and basidiomycetes. Our method uses only four reagents, namely TE buffer, KOH, (NH4)2SO4, and BSA, which are easily available in biology laboratories. It does not require specialized equipment. We found that both high and low copy number genes could be amplified using this method. In addition, we demonstrated its applicability to real-time PCR as well as the conventional PCR. Taken together, we expect that this method can accelerate PCR-based macrofungal research such as molecular identification, DNA marker typing, gene cloning, and transformant screening.
SUPPLEMENTARY MATERIALS
http://www.kjmycology.or.kr/article/?num=21-035_Supplementary_Materials


Fig. S2. Nucleotide sequence of MITOCHONDRIAL INTERMEDIATE PEPTIDASE (MIP) gene of Sanmaru 1ho. The nucleotide sequence of the PCR product amplified in Fig. 3b were determined by DNA sequencing and aligned with GeneBank sequence JN129270 using the EMBOSS Needle (https://ebi.ac.uk/Tools/psa/emboss_needle/).
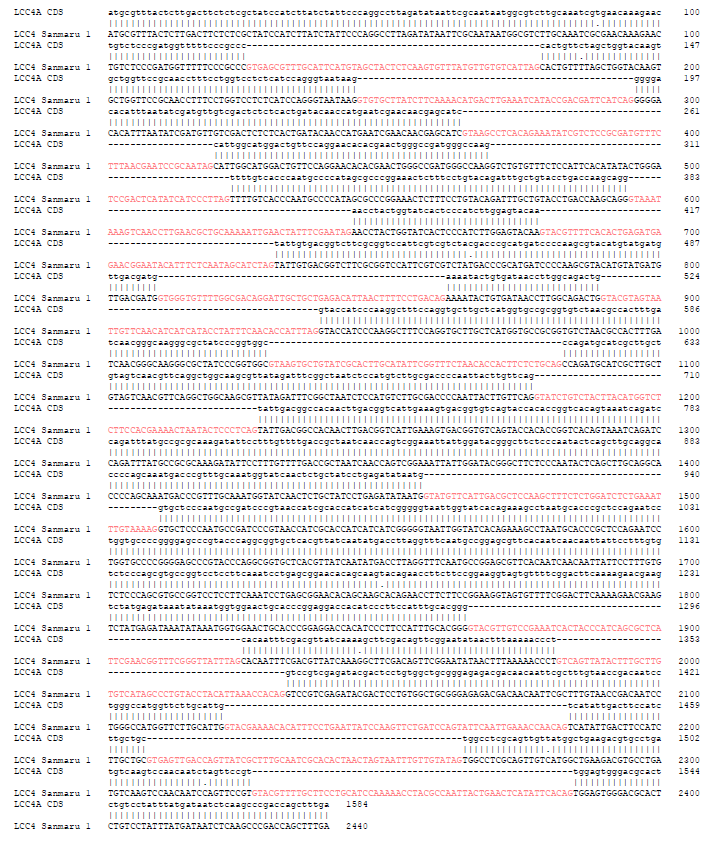
Fig. S3. Nucleotide sequence of LACCASE 4 (LCC4) gene of Sanmaru 1ho. The nucleotide sequence of the PCR product amplified in Fig. 3b were determined by DNA sequencing and aligned with GeneBank sequence JX879728 using the EMBOSS Needle (https://ebi.ac.uk/Tools/psa/emboss_needle/) with some modifications to follow GT-AG rule. Note that the predicted introns are indicated in pink.

Fig. S4. Nucleotide sequence of PHOTORECEPTOR A (PHRA) gene of Sanmaru 1ho. The nucleotide sequence of the PCR product amplified in Fig. 3b were determined by DNA sequencing and aligned with GeneBank sequence AB279630 using the EMBOSS Needle (https://ebi.ac.uk/ Tools/psa/emboss_needle/). Intron region is indicated in pink. Note that 2 amino acids are inserted in PHRA gene of Sanmaru 1 ho compared to the reference sequence.
ACKNOWLEDGEMENTS
This work was supported by the Golden Seed Project of ‘Breeding of new strains of shiitake for cultivar protection and substitution of import [213007-05-5-SBH10]’ provided by the Ministry of Agriculture, Food and Rural Affairs, Ministry of Oceans and Fisheries, Rural Development Administration and Korea Forest Service.



