서론
세계적으로 식량 소비가 증가함에 따라 버섯에 대한 수요도 함께 증가하고 있다[1]. 2022년의 국내 버섯생산량은 142,352 톤/년으로 기타 버섯(40.6%), 느타리버섯(35.1%), 팽이버섯(18.6%), 양송이버섯(5.6%), 영지버섯(0.07%)으로 보고가 되었으며, 2021년 기준보다 총 생산량 143,620 톤/년보다 다소 감소하였다[2]. 줄어든 생산량에 비하여 수확 후 배지의 발생은 꾸준히 유지되고 있는데, 버섯을 수확한 후 남은 배지를 수확 후 배지(spent mushroom substrate, SMS)라고 하며, 1 kg의 버섯을 생산하는데 약 5 kg의 수확 후 배지가 발생한다[3]. 또한 버섯 배지에 남아 있는 성분의 경우 버섯 재배 기간동안 약 15-25%만 영양원으로 활용이 되고, 나머지는 수확 후 배지로 배출되기에 다른 농업 부산물에 비하여 상당히 높은 유기물 함량을 지니고 있으며[4], 가축 사료화[5] 및 퇴비화[6] 같은 재활용 관련 연구가 지속되고 있지만 버섯 배지 제작하는 과정에서 수분, 원료, 배합비율, 재배방식이 지역과 농가마다 상이하기에 재활용할 수 있는 연구가 상당히 제한적이다. 그렇기에 현재 수확 후 배지는 적절한 공정 과정 없이 곧바로 농경지 퇴비처리로 폐기하고 있으며[7], 이러한 무분별한 퇴비 처리는 토양의 염류 집적과 수질오염, 악취 발생 및 온실가스 배출 같은 환경 문제를 유발한다[8]. 많은 연구자들은 2026년에는 전세계적으로 약 1억톤의 수확 후 배지가 발생할 것이라 예상하고 있는 만큼[9], 버섯 산업의 지속가능성과 농업 폐기물 재활용 그리고 농업의 경제 발전을 위해 수확 후 배지를 효과적으로 활용할 수 있는 연구가 필요한 실정이다[10]. 현재까지 여러 종류의 수확 후 배지를 대상으로 마이크로바이옴 분석을 진행하여 더 넓은 재활용 연구에 기반이 되는 자료는 미비하기에 본 연구에서는 버섯 배지에 첨가되는 재료에 따라 변하는 세균 군집도를 규명하고, 이를 통한 선택성 유기농자재로써 개발 가능성을 확인하기 위해 본 연구를 수행하였다. 먼저 3종의 표고, 느타리, 새송이버섯의 수확 후 배지를 이용하여 차세대 염기서열 분석기술(next generation sequencing, NGS)을 통해 버섯간의 세균 군집 비교 및 분석을 하였으며, 더불어 수확 후 배지간 세균 군집 유사도까지 확인하였다.
재료 및 방법
수확 후 배지의 시료 채취
본 연구를 위해 2023년 10월 1달간, 강원도 춘천, 홍천, 경기도 광주, 여주, 충청남도 아산, 경상남도 의령에 위치한 표고, 느타리, 새송이버섯 농가에 방문하여 수확 후 배지의 시료를 무작위로 10점씩 채취하였다. 채취된 시료 중 유일하게 병 재배가 아닌 봉지 재배 방식인 표고 수확 후 배지 같은 경우는 여러 균상 위치에서 10점씩 채취하였으며, 채취된 모든 시료는 깨끗한 클린백에 담은 후 개별 밀봉을 하였다. 그 후 아이스박스에 넣어 실험실로 옮긴 후 -4℃ 냉동고에 저장하면서 NGS 및 이화학적 분석을 진행하였다(Table 1). 더불어 6곳의 버섯 농가에서 재배된 배지 재료와 배합 비율은 다음과 같다(Table 2).
수확 후 배지의 NGS 분석
수확 후 배지 내의 마이크로바이옴 분석을 위하여 채취된 수확 후 배지를 멸균된 막자 사발과 스푼을 이용하여 곱게 마쇄한 후 각각의 처리구 별로 무작위 sampling을 진행하였다. 그리고 DNA 추출을 위해 수확 후 배지의 시료를 멸균된 buffer (NaCl 4.25 g, Na2 HPO4 0.3 g, MgSO4 0.1 g, KH2 PO4 0.15 g, 젤라틴 0.05 g)에 넣어 섞어준 후 10분간 정치하였다. 그리고 5분동안 4,000 rpm로 원심분리한 후 수확 후 배지의 용액을 얻었다. 각각의 수확 후 배지 용액은 FastDNA spin kit for soil (MP Biomedicals, California, USA)제품을 사용하여 추출 방법에 따라 추출하였다. 추출된 DNA sample은 ㈜마크로젠(Seoul, Korea)에 의뢰하여 NGS 분석을 진행하였다. Total DNA 추출에는 DNeasy PowerSoil Kit (Qiagen, Hilden, Germany)를 활용하여 제품의 manual에 따라 진행하였다. 추출된 Total DNA는 Quant-IT PicoGreen을 이용하여 정량화하였으며, 1차 PCR에 사용된 primer는 Illumina에서 제공하는 16S rDNA primer 서열(Illumina_16S_341F: 5’-TCGTCGGCAGCGTCAG ATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3'; Illumina_16S_805R: 5'-GTCTCGTGG GCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC-3')를 이용하였다[11]. 1차 PCR 산물은 AMPure beads (Agencourt Bioscience, Beverly, MA, USA)로 정제한 후, NexteraXT Indexed Primer를 사용하여 최종 library 구축을 위한 PCR 증폭을 진행하였다. 2차 PCR 조건은 10 cycle을 제외하고는[11] 조건과 동일하였으며, PCR 산물은 AMPure beads로 정제하였다. 이후 qPCR Quantification Protocol Guide (KAPA Library Quantification kits for Illumina sequencing platforms)에 따라 최종 정제된 산물을 정량화하였다. Illumina사에서 제공하는 MiSeq platform (Illumina, Sandiego, USA)을 활용하여 paired-end (2×300 bp) sequencing을 수행하였으며, 세균 군집 비교 분석을 위해 QIIME 프로그램[12]을 이용하여 sequencing 데이터 분석을 진행하였다.
수확 후 배지의 α-diversity (Amplicon Sequence Variants [ASV], Rarefaction, Chao1, Shannon, Gini-Simpson) 및 β-diversity 분석
Illumina MiSeq을 이용하여 Sequencing이 완료한 후, Cutadapt 프로그램[13]을 사용하여 sequencing adapter 서열과 target 유전자 영역의 primer 서열을 제거 후 Forward 서열(Read1)과 Reverse 서열(Read2)을 각각 250, 200 bp로 자르는 전처리 과정 진행하였다. 그리고 DADA2 package [14]를 사용하여 Amplicon sequencing 과정의 오류를 교정(error-correction)하였다. 마지막으로 Paired-end 서열을 하나의 서열로 조립한 뒤, DADA2의 Consensus method를 사용하여 Chimera 서열을 제거하고 ASVs를 형성하였고, 이때 최초 raw data (read counts)는 H-NT: 113,734, G-NT: 114,296, C-PG: 86,860, Y-PG: 103,968, U-SS: 93,898, A-SS: 102,318로 확인되었다. 이렇게 생성된 ASV 중 350 bp 미만의 ASV는 제외하였고 세균 군집 비교 분석을 위해 QIIME 프로그램을 이용하여 전체 샘플 중 최소 read 수를 기준으로 subsampling을 적용하여 normalization하였다. 최종적으로 검출된 ASVs abundance와 taxonomy 정보로 QIIME을 이용하여, Chao1, Shannon, GiniSimpson 및 Rarefaction 확인을 통하여 수확 후 배지내 세균 군집의 종 다양성 및 균등도를 분석하였으며, Weighted unifrac distance를 기반으로 샘플 간 beta diversity 값을 구하고, WPGMA (Weighted Pair Group Method with Arithmetic Mean) tree [15]를 통해 샘플 간 유연관계를 시각화 하였다.
결과 및 고찰
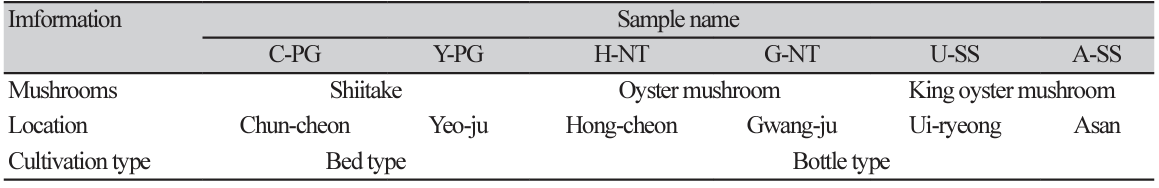
수확 후 배지의 ASV 분석
ASV 분석 결과, 느타리버섯의 ASV 지수가 다른 수확 후 배지에 비하여 높은 값이 관찰되었으며 이는 느타리버섯의 수확 후 배지가 세균 다양성이 높음을 의미한다(Fig. 1). 더불어 NT (느타리)>PG (표고)>SS (새송이) 수확 후 배지 순서로 ASV 값을 나타났는데 H-NT와 G-NT처리구를 제외한 나머지 C-PG, Y-PG 그리고 U-SS, A-SS처리구간이 비슷한 세균 다양성을 보였다. 실제로 Sun [16]등은 미생물 군집 구성에 대한 중요한 부분은 토양의 유기화학적 성분과 pH라고 말하고 있으며, 연구 결과를 따르면 작물 부산물인 밀짚과, 가축 분뇨(소, 돼지)를 각각 황폐화된 토양에 첨가한 후 마이크로바이옴 분석을 진행하였을 때, 밀짚을 혼합한 토양에서는 세균 군집이 풍부해진 반면, 가축 분뇨를 첨가한 토양에서는 군집이 무처리 토양과 비슷하거나, 더 낮은 군집을 보였다. 이처럼 본 ASV분석 결과도 버섯 재배 지역은 다르지만, 버섯 종류마다의 배지에 첨가되는 재료가 유사하기에 종 다양성이 비슷한 수치가 나온 것으로 판단된다.
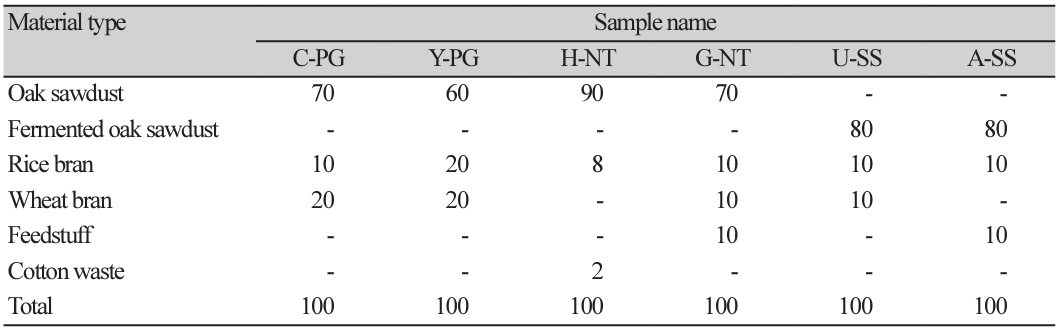
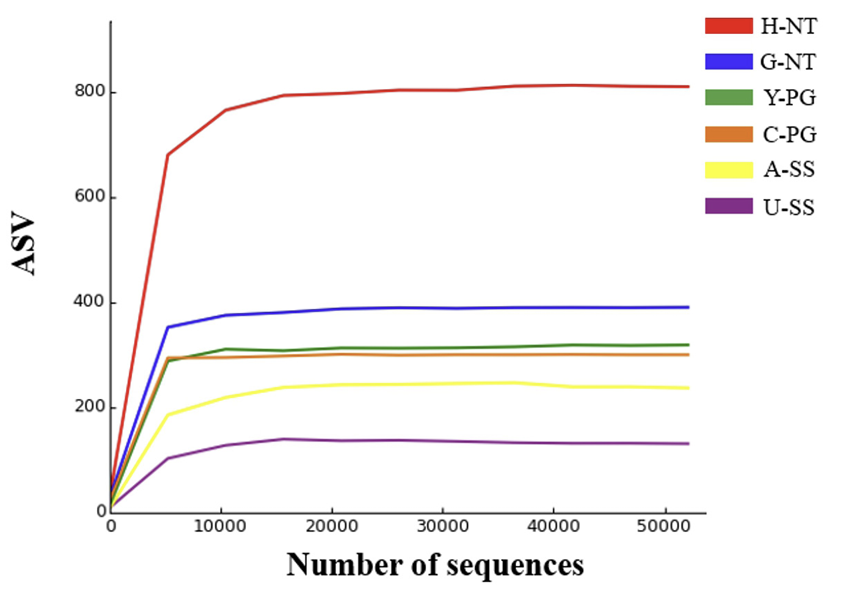
수확 후 배지의 α-diversity (Rarefaction, Chao1, Shannon, Gini-Simpson) 분석
ASV 값을 기준으로 α-diversity인 Rarefaction, Chao1, Shannon, Gini-Simpson를 분석하였다. Rarefaction은 처리구의 sampling 과정에서 나타나는 종의 대한 다양성을 평가 및 계산할 수 있고, Rarefaction curve를 제작한 후 curve가 직선으로 접어들면 처리구 내의 종들이 대부분 검출되었다는 것이며, 반대로 꾸준히 상승한다면, 아직 검출되지 않은 많은 종이 있다는 것을 의미하기에 미검출 종의 존재 가능성 여부도 추정할 수 있다[17]. 본 연구에 사용된 6개의 수확 후 배지 처리구의 rarefaction curve에서는 NT>PG>SS 순서로 다양성을 보였다. 특히 H-NT는 독보적으로 높은 종 풍부도를 보였는데, 다른 처리구들에 비하여 직선에 접어드는 시간이 상대적으로 늦었기에 검출되지 않은 미검출 종도 다수 있음을 시사한다(Fig. 2). 또 다른 α-diversity인 Chao1, Shannon index은 수치가 클수록 미생물의 종 다양성이 높은 것을 나타내는데[18], 이 결과도 ASV 값과 동일하게 느타리버섯 수확 후 배지 내의 Chao1, Shannon지수가 처리구 중 가장 높은 수치로 나타났다(Table 3). 또한 Gini Simpson index는 0에 가까울수록 다양성이 낮은 것을 나타내고, 반대로 1에 가까울수록 다양성이 높은 것을 의미한다[19]. Gini-simpson index도 느타리버섯 수확 후 배지 처리구가 1에 가장 가까운 것으로 나타났으며, 특히 홍천에 위치한 느타리버섯 농가 H-NT처리구는 ASV를 포함한 Chao1, Shannon, Gini-Simpson index가 공통적으로 높은 수치로 나온 이유를 짐작하자면, H-NT처리구 버섯 배지의 배합비는 참나무 생톱밥:폐면:쌀겨(90:2:8)로 제작되었는데, 다른 버섯 배지에 들어가지 않은 폐면이 혼합되어 있었다. 대부분 느타리버섯 농가 중 호남권과 충청도 일부 지역, 벼 재배를 하는 지역을 제외하고는 대부분 폐면을 사용하고 있는데[20], 실제로 버섯 배지 재료에 들어가는 폐면, 활엽수 톱밥, 미강의 세균 수를 측정한 결과를 보면 미강이 가장 적었으며, 톱밥 그리고 폐면 순서로 세균 수가 많았다[21]. 본 실험 결과처럼 폐면이 혼합되어 있는 H-NT처리구가 다른 처리구에 비하여 세균의 다양성이 높은 이유는 폐면 혼합의 유무라고 판단된다.
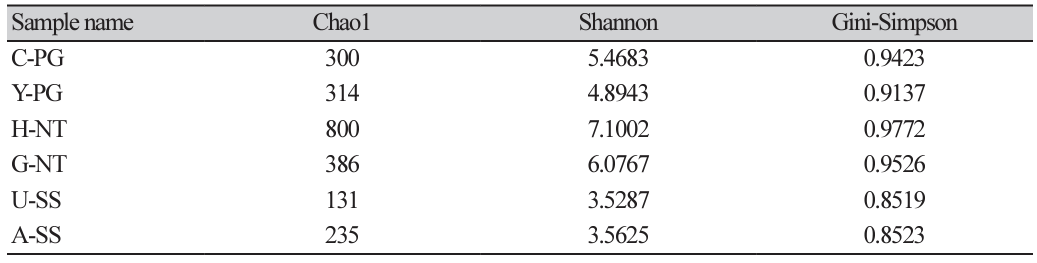
수확 후 배지의 WPGMA 분석
세균 군집 유사도를 확인하기위해 처리구간 비교하여 계산된 β-diversity 값을 이용하여 tree로 제작하였으며(Table 4), 각 처리구마다 수확 후 배지내 존재하는 세균의 phylogenetic diversity를 나타내었다. Lozupone [22,23]은 두 군집의 계통에 따라 유의하게 차이가 있는지 분석하기 위해 Unifrac 및 Weighted unifrac이라는 새로운 통계 방법을 제안하였다. 위 방법은 세균을 포함한 미생물의 군집을 비교하기 위해 최근 수많은 연구에 널리 적용되었으며, dendrogram의 거리가 짧을수록 처리구간 미생물 군집의 유사도가 높음을 의미한다[24]. WPGMA tree의 sample간 세균 군집도를 비교한 결과, 여러 종류 별 수확 후 배지간의 세균 군집이 서로 비슷한 것을 알 수 있다(Fig. 3). Jenkins [25]등은 연구에서는 가공하지 않은 바이오차를 영국, 이탈리아, 프랑스 토양에 혼합하여 개량한 후 마이크로바이옴 분석을 진행하여 미생물의 다양성을 확인하였는데, 3개의 국가 토양에 미생물 군집 분포도가 비슷하게 검출되었다. 이러한 결과를 빗대어 본 결과를 해석하자면 각 지역 버섯 농가에서 수집된 수확 후 배지는 지역 간의 지리적, 기후적 차이에 기인한 것이 아닌, 버섯 종류에 맞춰 사용된 배지 원재료의 유사성이 생물 군집 형성과 상관관계가 있는 것으로 판단된다. 더불어 보다 다양한 지역에서의 수확 후 배지를 수집하여 추가적인 분석이 필요하다고 사료된다.
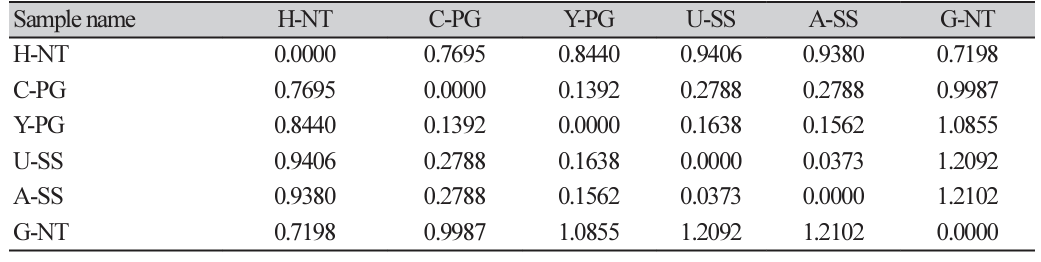
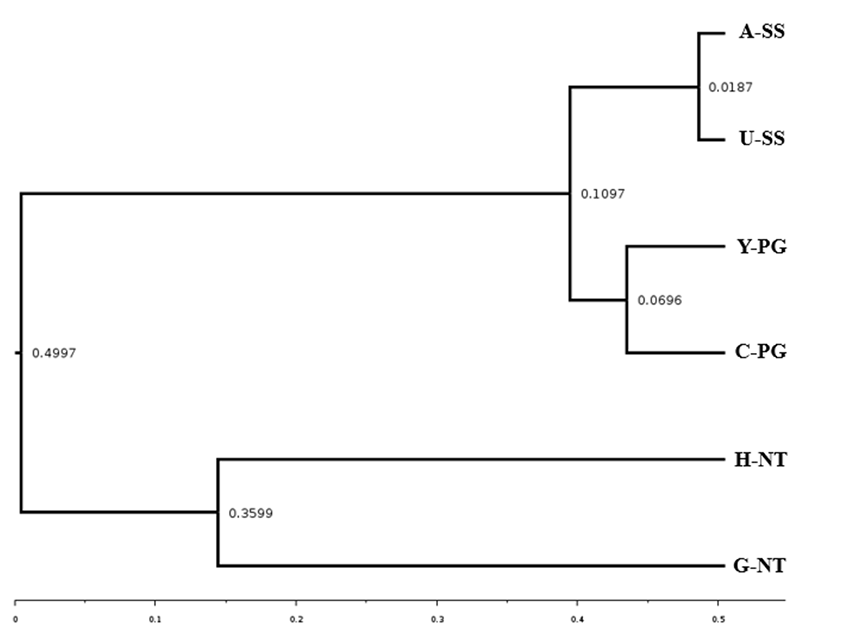
수확 후 배지의 문(Phylum) 수준 상대적 비교 분석
Phylum level에서 unclassified를 제외한, 전체 처리구에서 총 20 phylum level이 분류되었으며, 그중 처리구마다의 상위 7개 phylum level들을 재분류한 결과, Pseudomonadota, Bacteroidota, Bacillota, Actinomycetota, Acidobacteriota, Verrucomicrobiota, Thermomicrobiota로 나타났다(Table 5). 가장 높은 분포를 보인 Pseudomonadota는 사용 가능한 영양소가 많은 토양에서 번성하는 phylum으로 알려져 있다[26]. 특히 C-PG, Y-PG과 U-SS, A-SS 처리구에서 Pseudomonadota phylum이 87% 이상 높은 군집을 보였는데, C-PG와 Y-PG의 배지 제작 조성을 살펴보면 다른 처리구 배지 제작 조성보다 쌀기울, 밀기울의 배합비가 높은 것을 알 수 있다. 더불어 U-SS, A-SS처리구는 발효된 톱밥을 이용하여 배지를 제작했다는 공통점이 있는데, 발효톱밥에서 재배된 버섯이 높은 수확량과 직결된다는 연구 결과처럼[27], 일반 톱밥에 비하여 높은 영양분이 있기에 C-PG, Y-PG과 U-SS, A-SS 처리구에서 공통적으로 Pseudomonadota phylum이 높은 군집도를 보인 것으로 판단된다. 또한 느타리버섯의 수확 후 배지는 다른 처리구의 수확 후 배지에 비하여 cellulose, lignin 등과 같은 물질이 풍부하다고 알려져 있기에[28], 느타리버섯 처리구인 H-NT, G-NT에서 cellulose 분해에 관여하는 Bacteroidota와 Bacillota가 각각 높은 비율의 군집을 보였다[29]. 특히 H-NT처리구에서 Actinomycetota이 유일하게 높은 비율로 검출이 되었는데, Actinomycetota는 cellulose와 lignin 생분해에 중요한 역할을 하는 것으로 알려져 있으며[30], 실제로 H-NT처리구는 배지 제작 조성에 폐면이 혼합되었기에 섬유 분해 관련 세균의 군집이 상당수로 증가된 것이라 판단된다.
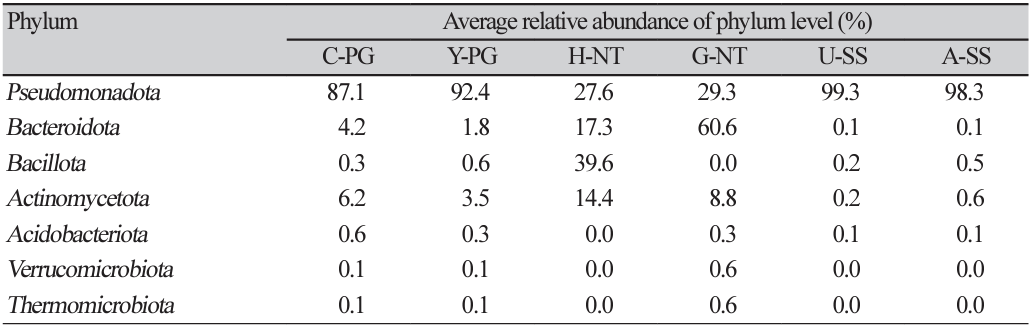
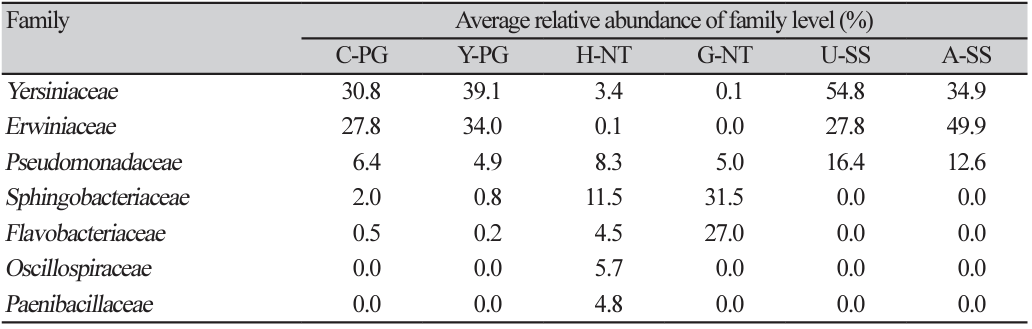
수확 후 배지의 과(Family) 수준 상대적 비교 분석
Family level에서 unclassified를 제외한, 전체 처리구에서 총 215 family level이 분류되었으며, 그중 처리구마다의 상위 7개 family level들을 재분류한 결과, Yersiniaceae, Erwiniaceae, Pseudomonadaceae, Sphingobacteriaceae, Flavobacteriaceae, Oscillospiraceae, Paenibacillaceae로 나타났다. 가장 다양하게 세균 군집이 분포되어 있는 처리구는 PG (표고버섯 수확 후 배지)와 H-NT (느타리버섯 수확 후 배지)였으며, SS (새송이버섯 수확 후 배지)처리구는 다양한 분포보다 집약적인 분포를 보였다(Table 6). G-NT처리구에서는 Flavobacteriaceae가 상대적으로 높은 비율을 보였는데, Flavobacteriaceae는 식물의 초기 생장에 긍정적으로 관여하며, poteinases나 chitinases와 같은 효소를 분비함과 동시에 항생제 같은 2차 대사물질을 분비한다고 알려져 있다[31]. 또한 H-NT처리구에서는 다른 처리구에서 검출되지 않은 Oscillospiraceae와 Paenibacillaceae이 검출되었는데 이들은 cellulase, hemicellulase 등과 같은 효소를 분비한다[32]는 공통적인 특징이 있으며, 유일하게 H-NT처리구에서만 검출된 이유는 배지 조성에 폐면이 혼합되었기에 나타난 결과임을 시사한다.
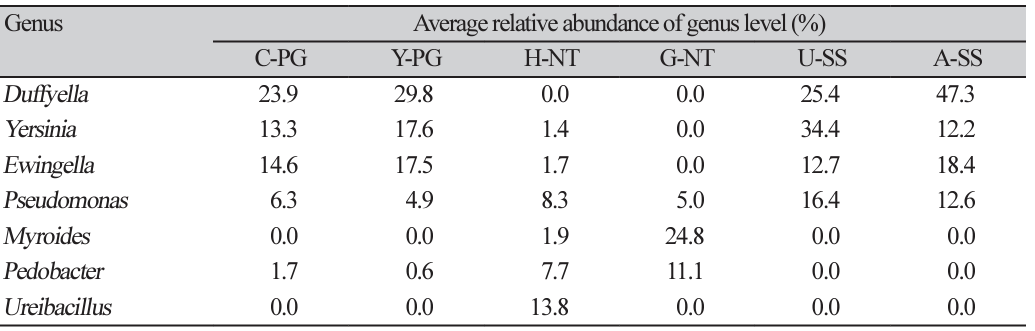
수확 후 배지의 속(Genus) 수준 상대적 비교 분석
Genus level의 전체 처리구에서 총 526 genus level이 분류되었으며, 그중 공통적으로 많이 분포된 상위 7개의 genus level들을 재분류한 결과, Duffyella, Yersinia, Ewingella, Pseudomonas, Myroides, Pedobacter, Ureibacillus로 나타났다. C-PG, Y-PG, U-SS, A-SS 처리구에서의 분포 비율은 과(Family)수준 상대적 비교 분석과 유사한 결과를 보였는데, PG와 SS 처리구에서 검출된 Duffyella, Yersinia, Ewingella는 느타리버섯 수확 후 배지인 H-NT, G-NT 처리구에서 검출되지 않거나, 상대적으로 낮은 비율로 확인되었으며, 반대로 H, G-NT 처리구에서 검출된 Pedobacter 및 Ureibacillus는 PG와 SS 처리구에서 검출되지 않거나, 분포 비율이 매우 적었다(Table 7). PG, SS 처리구에서 많은 분포를 나타낸 Duffyella는 아직 특성 연구가 미비한 실정이며, 최근 연구된 바로는 식물체 내의 이눌린을 비롯한 다양한 기능성 성분을 증가된다는 결과가 있었다[33]. 더불어 Yersinia는 사람에게 독성이 있으며[34], 토양에서 전염병을 유발하는 유해 세균으로 알려져 있는 것[35]을 보면 크게 버섯과 연관성이 없다고 판단할 수 있지만 추후 Duffyella과 Yersinia를 이용하여 버섯의 종균과 연관성이 있는지 확인할 필요가 있다. 또한 H-NT처리구에서 Ureibacillus의 분포 비율이 굉장히 높게 나타났는데, 이는 폐면을 사용한 느타리버섯 수확 후 배지에서 Ureibacillus이 검출된 연구와 동일한 결과를 보였으며[36], 폐면을 사용한 배지와 사용하지 않은 배지의 생산성을 비교하여 Ureibacillus의 유무에 따른 느타리버섯 생산성에 대한 연구가 필요하다고 판단된다.
적요
버섯 배지에 첨가되는 재료에 따라 변하는 수확 후 배지내의 세균 군집도를 확인하여 더 넓은 재활용 연구에 기여하고자 춘천, 여주, 홍천, 광주, 의령, 아산에서 수집한 표고, 느타리, 새송이버섯의 수확 후 배지를 대상으로 차세대 염기서열 분석을 진행하였다. ASV 값을 기반으로 α-diversity인 Rarefaction, Chao1, Shannon, Gini-Simpson를 분석한 결과, 유일하게 폐면이 혼합되어 있는 느타리버섯 수확 후 배지인 H-NT 처리구의 세균 다양성이 매우 풍부하였다. 또한 β-diversity의 WPGMA를 이용하여 처리구간 세균 군집의 유사성을 분석한 결과, 버섯 종류에 따라 유연관계가 가까운 것을 확인하였다. 본 연구를 시작으로 작물 및 토양에 유용한 특정 세균 비율이 높게 분포하고 있는 수확 후 배지를 연구한다면 맞춤형 유기농자재로서의 활용 가능성이 있음을 시사한다.

