INTRODUCTION
Apple (Malus pulmina) is a Rosaceae plant whose fruit is consumed worldwide fresh or processed. In Korea, apples are representative fruit trees. In 2018, the cultivation area of apples was 33,234 ha, accounting for 21.2% fruit tree cultivation area [1]. Recently, fire blight has damaged many apple trees in orchards across several provinces in Korea [2]. It is caused by Erwinia amylovora, a bacterium belonging to the Enterobacteriaceae [2]. According to the burial control regulations, fire-blighted apple trees are buried 4-5 m below the soil [3]. To ensure burial control safety, soil microorganisms in apple orchards must be explored to understand the environment of the burial soil.
Apple trees buried in the soil are mostly degraded and deteriorated by saprophytic soil fungi [4]. Thus, information on soil fungal diversity is important to understand the decay process of fire-blight-controlled apple trees. Saprophytic fungi produce enzymes that decompose cellulose, hemicellulose, and pectin present in plant materials. We investigated the fungal diversity in orchard soil where fire-blighted apple trees are buried. In this study, we report the description and morphological features of seven ascomycetes previously unreported in Korea.
MATERIALS AND METHODS
Soil sampling
Soil samples were collected by an excavator from three locations at each site in several apple orchards in Chungju where all diseased trees were buried for at least 24 months. Soil was collected with permission from the Rural Development Administration, Korea and in compliance with plant quarantine regulations. Approximately 300 g soil samples were collected using a sterile spade at approximately 3 m depth from the surrounding areas that had come into contact with the buried plants. The collected soil samples were placed in zipper bags in a low-temperature cooler and transported to the laboratory.
Isolation and morphological studies of fungi
The collected soil samples were analyzed in a biosafety level 2 licensed facility at the Dankook University. A portion of the soil sample was placed in a 50 mL sterile conical tube containing 20 mL sterile distilled water and then vortexed for 3 min to suspend the soil sample. The suspension was gradually diluted to 104 using sterile distilled water and 100 μL diluent was spread on Dichloran-Glycerol 18% (DG18) agar medium and cultured at 25℃ for 5 days. The fungal mycelia were transferred to new media, and fungal flora that appeared different from each other were selected for identification. The selected fungal strains were assigned a Dankook University Culture Collection (DUCC) number and grown for 7-9 days on potato dextrose agar (PDA), malt extract agar (MEA), Czapek yeast agar (CYA), and oatmeal agar (OA) (BD Science, Franklin Lakes, USA) for morphological studies. The colony size, shape, and color were observed using a stereomicroscope (BX53; Olympus, Tokyo, Japan). Mycelial growth was recorded by measuring the colony diameters. The microstructures were observed using a phase-contrast microscope (Karl Zeiss, Axioskop 40). For scanning electron microscopy (SEM), the fungal isolate was grown for 5-7 days at 25℃ on 2% PDA plates overlaid with cellophane (Bio-Rad Laboratories, Hercules, Canada) and the ultrastructures were examined with a Hitachi S-4300 scanning electron microscope operating at 15 kV [5].
Molecular analysis
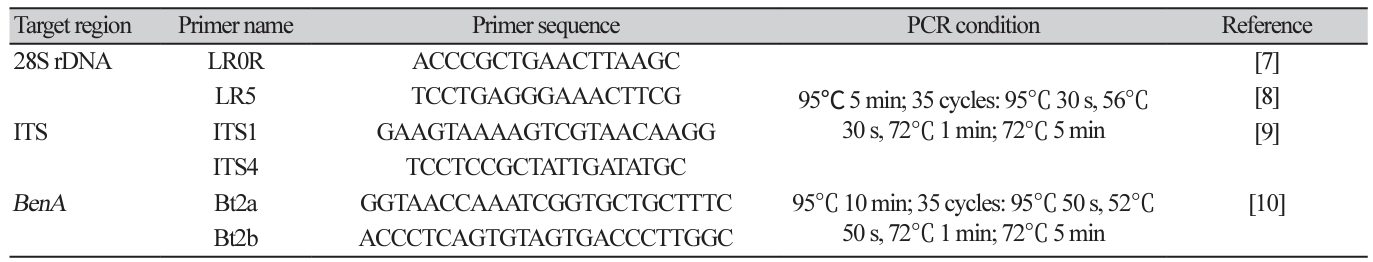
For molecular analysis, fungi were grown on PDA for 5 days at room temperature, and fungal genomic DNA was extracted from the cultured fungal mycelia using a Direct DNA Prep kit (Navibiotech, Cheonan, Korea). Using the extracted DNA as a template, molecular taxonomic marker sequences were amplified by PCR (T100 Thermal Cycler; BIO-RAD, Hercules, USA) using the primers for ITS1-5.8S-ITS2 region (ITS region), 28S large subunit of the nuclear ribosomal RNA gene (LSU region), and partial sequences of the β-tubulin, calmodulin, and translation elongation factor 1-α genes (Bioneer Corp., Daejeon, Korea; Table 1) [6-12]. The PCR conditions are listed in Table 1. PCR amplicons were electrophoresed on 1% agarose gel to confirm the amplified DNA bands. The confirmed PCR amplicon was purified using the High Pure PCR Product Purification Kit (Roche, Indianapolis, USA) and submitted to Macrogen (Seoul, Korea) for determining the nucleotide sequence.
The fungal homologs for nucleotide sequences were searched in the NCBI DB using the BLAST program at the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov). The reference sequences were obtained from the NCBI GenBank DNA database (Table 2) and analyzed using the ClustalW alignment tool of the MEGA 11 program to obtain a phylogenetic tree for the phylogenetic analysis [13-14]. The maximum likelihood method was used to construct the phylogenetic tree based on aligned sequences [15]. The Kimura 2-parameter model was used to estimate genetic differences [16]. The reliability of the nodes in the phylogenetic tree was confirmed using 1,000 bootstraps.
Table 2
The fungal species used to construct the phylogenetic tree and GenBank accession numbers of the five marker sequences for phylogenetic analysis(continued).

Table 2
The fungal species used to construct the phylogenetic tree and GenBank accession numbers of the five marker sequences for phylogenetic analysis.(continued)


RESULTS AND DISCUSSION
The morphological characteristics and the results of the molecular phylogenetic analysis of the seven unrecorded soil fungi are described below. The nucleotide sequences of these seven DUCC fungal strains were registered in the GenBank database, and the accession numbers are listed in Table 1. The strains were deposited at the National Institute of Biological Resources of the Republic of Korea and received NIBRFGC numbers.
Aspergillus aureolatus
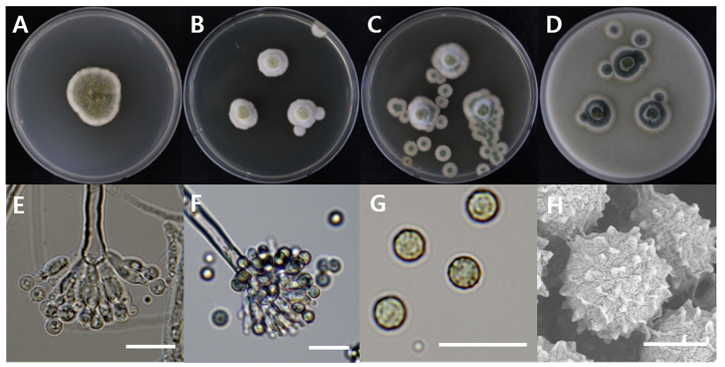
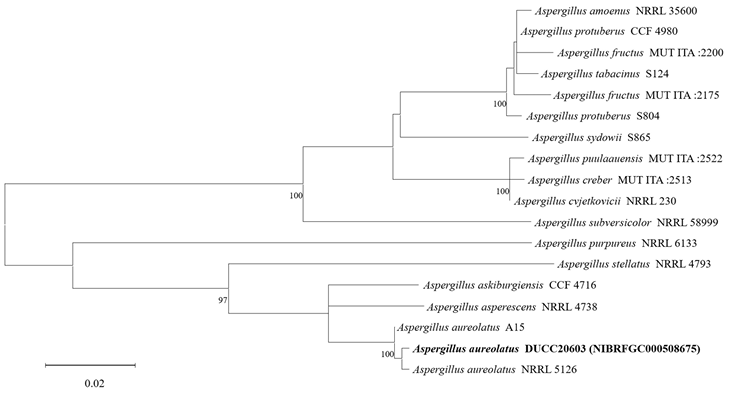
The diameters of Aspergillus aureolatus DUCC20603 (NIBRFGC000508675) colonies were 28 mm on PDA after 14 days and 11, 12, and 14 mm on MEA, CYA, OA, respectively, after 7 days. The colonies were olive green with a white border, white, green with a thick white border, and dark green with a thin white border on PDA, MEA, CYA, and OA, respectively (Fig. 1). The DUCC20603 strain has 3-4 μm spherical conidia and 5-6 μm elongated cylindrical phialide. When observed under SEM, the conidia had projections on the surface (Fig. 1). Based on the morphological characteristics and molecular phylogenetic analysis, the DUCC20603 strain was identified as Aspergillus aureolatus, showing 99.84% calmodulin gene sequence similarity to that of A. aureolatus (GenBank accession no. LR593496.1; Fig. 2; Table 2). A. aureolatus was isolated from air and reported in Yugoslavia in 1964 [17].
Fig. 1
Colony morphology of Aspergillus aureolatus DUCC20603 (NIBRFGC000508675) strain grown on different media and their microstructures. A: Potato dextrose agar (PDA). B: Malt extract agar (MEA). C: Czapek yeast agar (CYA). D: Oatmeal agar (OA). E-G: Light microscopic images of conidia, phialides, metulae, vesicle, and conidiophores. H: Scanning electron microscopy (SEM) image of conidia. Scale bar, E-G=10 µm, H=2 µm.
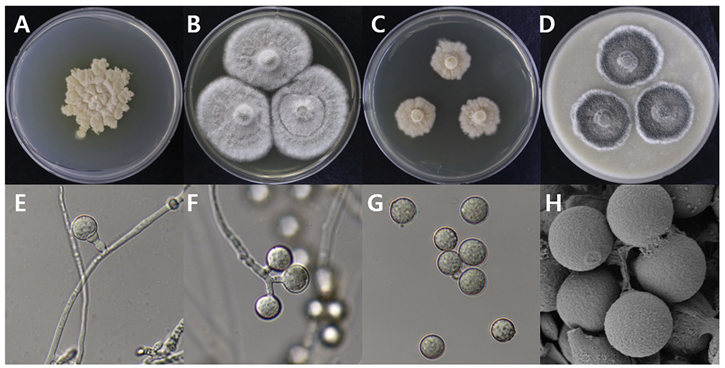
Botryotrichum atrogriseum
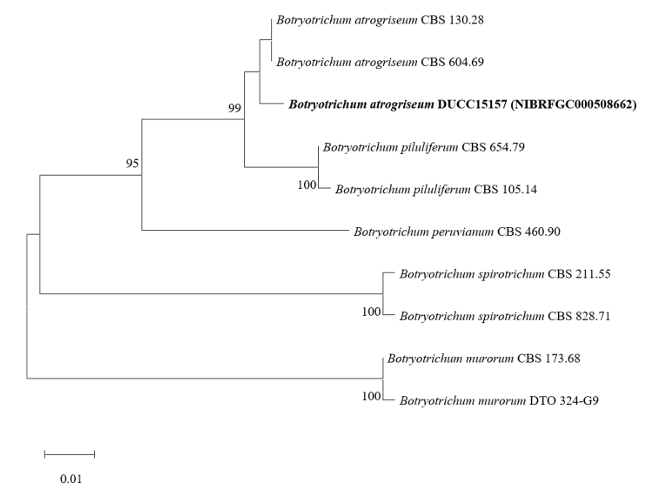
Botryotrichum atrogriseum DUCC15157 (NIBRFGC000508662) colonies grew to 34, 45, 12, and 22 mm on PDA, MEA, CYA, and OA, respectively, after 14 days. The colonies were pale yellow, white, light yellow, and gray with a white border on PDA, MEA, CYA, and OA, respectively (Fig. 3). The DUCC15157 strain has ≥130 μm diameter conidiophores and 21-25 μm light brown spherical conidia (Fig. 3). Based on the morphological characteristics and molecular phylogenetic analysis, the DUCC15157 strain was identified as Botryotrichum atrogriseum, showing 99.51% β-tubulin gene sequence similarity with that of B. atrogriseum (GenBank accession no. KX976932.1; Fig. 4; Table 2). B. atrogriseum was first reported in 1929 and may be used as a biological control agent against Fusarium wilt [18-19].
Fig. 3
Colony morphology of Botryotrichum atrogriseum DUCC15157 (NIBRFGC000508662) strain grown on different media and their microstructures. A: Potato dextrose agar (PDA). B: Malt extract agar (MEA). C: Czapek yeast agar (CYA). D: Oatmeal agar (OA). E-G: Light microscopic images of conidia and conidiophore. H: Scanning electron microscopy (SEM) image of conidia. Scale bar, E-G=10 µm, H=2 µm.
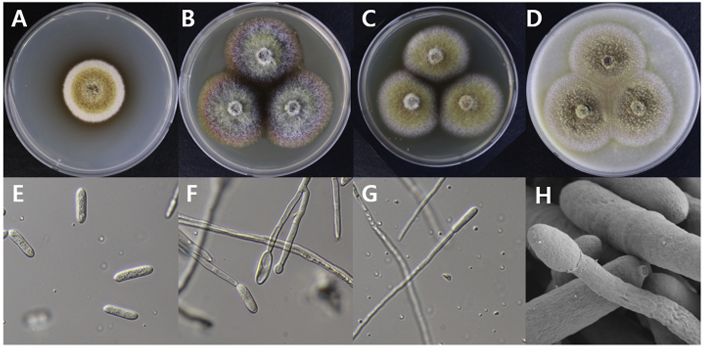
Dactylonectria novozelandica
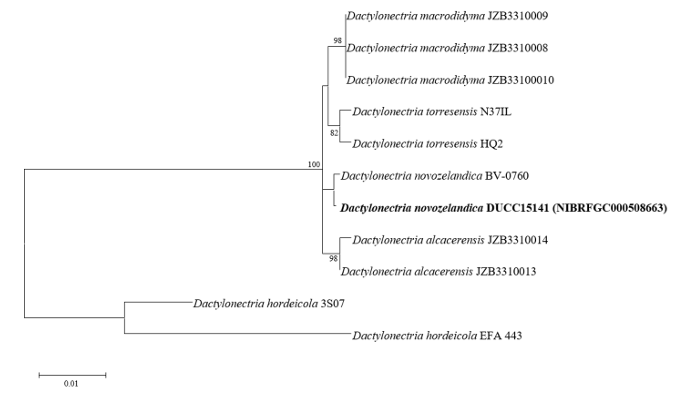
Dactylonectria novozelandica DUCC15141 (NIBRFGC000508663) colonies grew to 23, 25, 19 and 21 mm in diameter on PDA, MEA, CYA, and OA, respectively, after 7 days. The colonies were yellow with a thick white border on PDA and CYA and brownish yellow with a white border on OA (Fig. 5). From the center, the colony color was yellow, purple, or brown on MEA. The DUCC15141 strain produced a brown secondary metabolite in all four media. The DUCC15141 strain has 40-100 μm conidiophores and 3.54.0×25-30 μm² rod-shaped conidia (Fig. 5). Based on the morphological characteristics and molecular phylogenetic analysis, the DUCC15141 strain was identified as Dactylonectria novozelandica, showing 99.65% translation elongation factor 1 alpha (TEF-1α) sequence similarity with that of D. novozelandica (GenBank accession no. MK602815.1; Fig. 6; Table 2). D. novozelandica is the basionym of Ilyonectria novozelandica and was isolated from Vitis vinifera in 2012 [20]. It also causes root and crown rot of strawberries in Uruguay [21].
Fig. 5
Colony morphology of Dactylonectria novozelandica DUCC15141 (NIBRFGC000508663) strain grown on different media and their microstructures. A: Potato dextrose agar (PDA). B: Malt extract agar (MEA). C: Czapek yeast agar (CYA). D: Oatmeal agar (OA). E-G: Light microscopic images of conidia and conidiophore. H: Scanning electron microscopy (SEM) image of conidia and conidiophores. Scale bar, E-G=10 µm, H=2 µm.
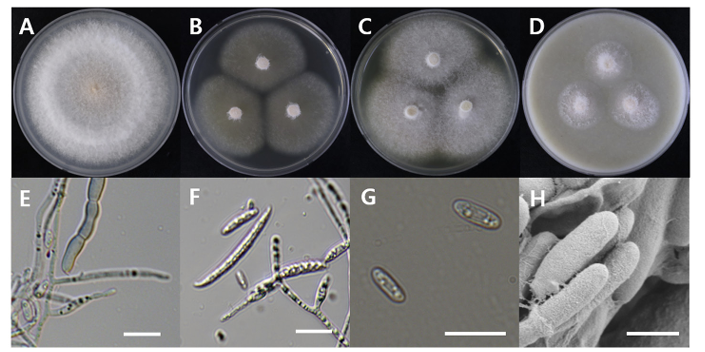
Fusarium denticulatum
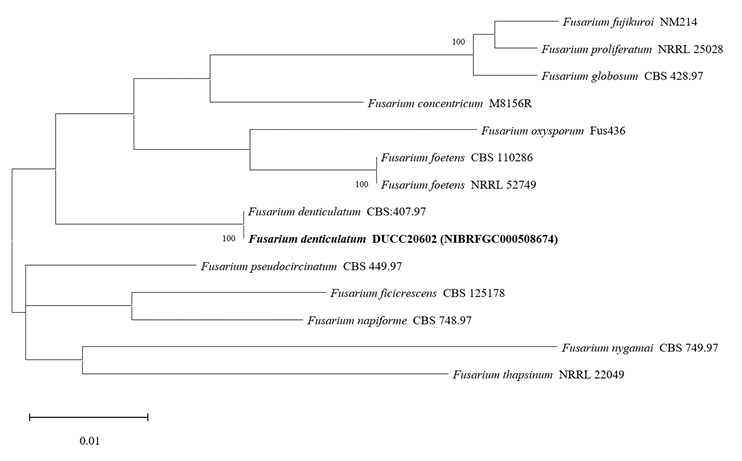
Fusarium denticulatum DUCC20602 (NIBRFGC000508674) colonies grew to 47, 45, 47 and 21 mm on PDA, MEA, CYA, and OA, respectively, after 7 days. The colonies were white with mycelial band edges on PDA, light orange on MEA, and white on CYA and OA (Fig. 7). The DUCC20602 strain has 6-8 µm elliptical conidia (Fig. 7). Based on the morphological characteristics and molecular phylogenetic analysis, DUCC20602 strain was identified as Fusarium denticulatum, showing 99.78% TEF-1α sequence similarity with that of F. denticulatum (GenBank accession no. MT011002.1; Fig. 8; Table 2). F. denticulatum was isolated from Ipomoea batatas in Louisiana in 1998 and caused chlorotic leaf distortion in sweet potatoes in China [22-23].
Fig. 7
Colony morphology of Fusarium denticulatum DUCC20602 (NIBRFGC000508674) strain grown on different media and their microstructures. A: Potato dextrose agar (PDA). B: Malt extract agar (MEA). C: Czapek yeast agar (CYA). D: Oatmeal agar (OA). E-G: Light microscopic images of conidia, conidiophore, and macroconidia. H: Scanning electron microscopy (SEM) image of conidia and conidiophores. Scale bar, E-G=10 µm, H=1 µm.
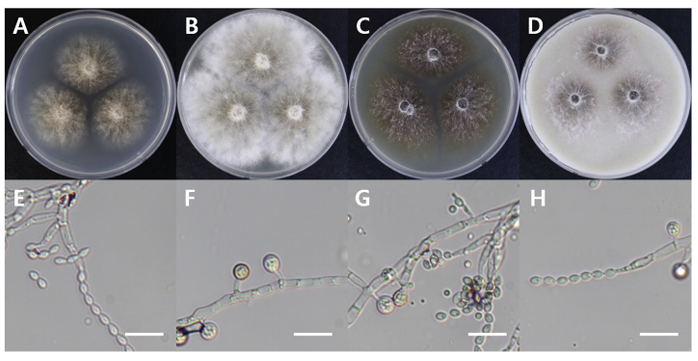
Paecilomyces tabacinus
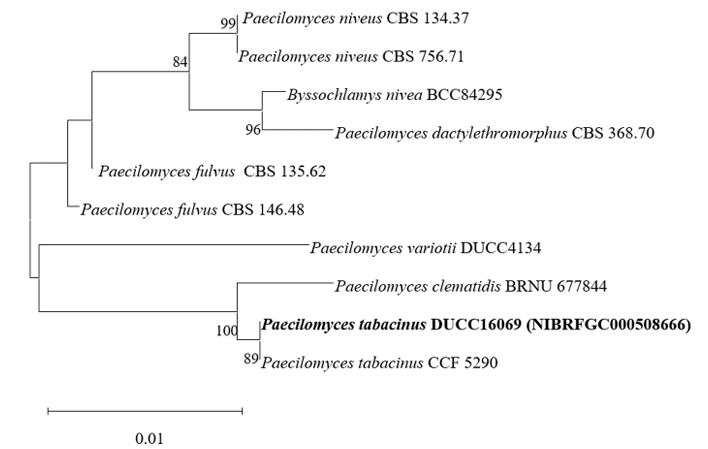
Paecilomyces tabacinus DUCC16069 (NIBRFGC000508666) colonies grew to 35, 55, 39, and 41 mm on PDA, MEA, CYA, and OA, respectively, after 7 days. The colonies were light brown on PDA, white with a brown center on MEA and OA, and dark brown on CYA. The DUCC16069 strain has 2-6 µm oval or globular conidia (Fig. 9). Based on the morphological characteristics and molecular phylogenetic analysis of the LSU region, DUCC16069 strain was identified as Paecilomyces tabacinus, showing 100 region sequence similarity with that of P. tabacinus (GenBank accession no. LT548280.1; Fig. 10; Table 2). P. tabacinus was isolated from the leaves of Nicotiana tabacum in North Carolina, USA in 2016 [24].
Fig. 9
Colony morphology of Paecilomyces tabacinus DUCC16069 (NIBRFGC000508666) strain grown on different media and their microstructures. A: Potato dextrose agar (PDA). B: Malt extract agar (MEA). C: Czapek yeast agar (CYA). D: Oatmeal agar (OA). E-H: Light microscopic images of conidia and conidiophores. Scale bar, 10 µm.
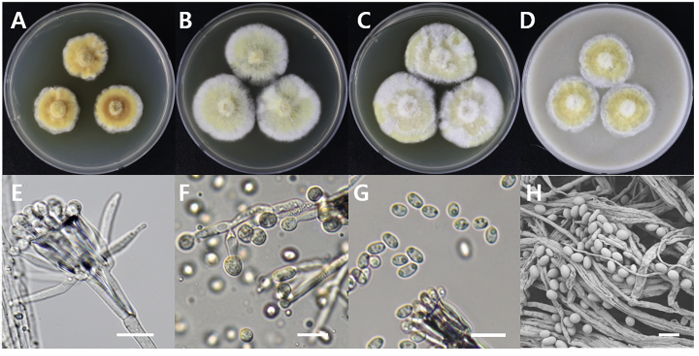
Sarcopodium tibetense
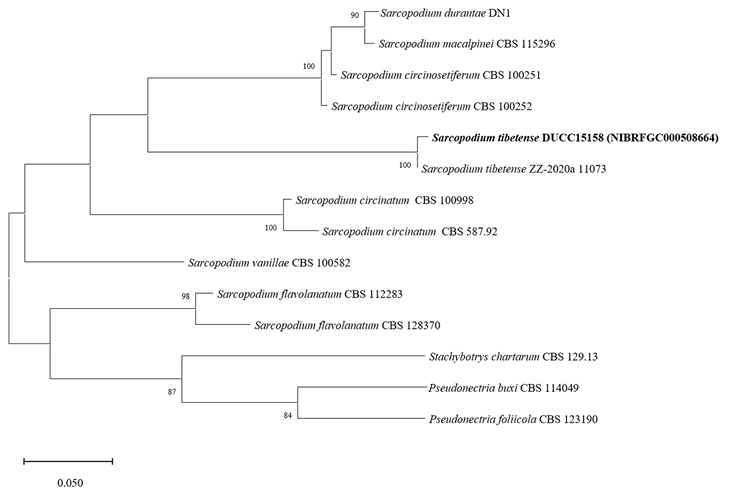
Sarcopodium tibetense DUCC15158 (NIBRFGC000508664) colonies grew to 21, 35, 36, and 23 mm on PDA, MEA, CYA, and OA, respectively, after 7 days. The colonies were dark yellow, pale yellow with a white border, mixed yellow and white, and yellow with a white border on PDA, MEA, CYA, and OA, respectively (Fig. 11). The DUCC15158 strain produces 1-1.5×3.5-4.0 µm² elliptical conidia from very dense (10 µm or more overlapped) and short (10-15 µm) as well as 4-5 μm circular conidia from undifferentiated conidiophores. When observed using SEM, the conidia appeared as slightly oval-like eggs (Fig. 11). Based on the morphological characteristics and molecular phylogenetic analysis of the β-tubulin gene, DUCC15158 was identified as Sarcopodium tibetense, showing 99.40% β-tubulin sequence similarity with that of S. tibetense (GenBank accession no. MT648557.1; Fig. 12; Table 2). S. tibetense was reported from China in 2021 [25].
Fig. 11
Colony morphology of Sarcopodium tibetense DUCC15158 (NIBRFGC000508664) strain grown on different media and their microstructures. A: Potato dextrose agar (PDA). B: Malt extract agar (MEA). C: Czapek yeast agar (CYA). D: Oatmeal agar (OA). E-G: Light microscopic images of conidia and conidiophore. H: Scanning electron microscopy (SEM) image of conidia and conidiophores. Scale bar, E-G=10 µm, H=2 µm.
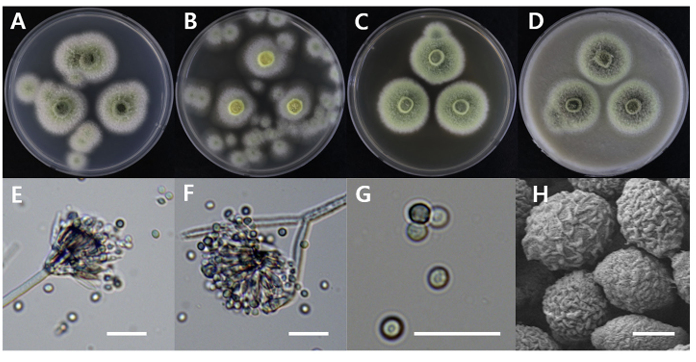
Talaromyces stollii
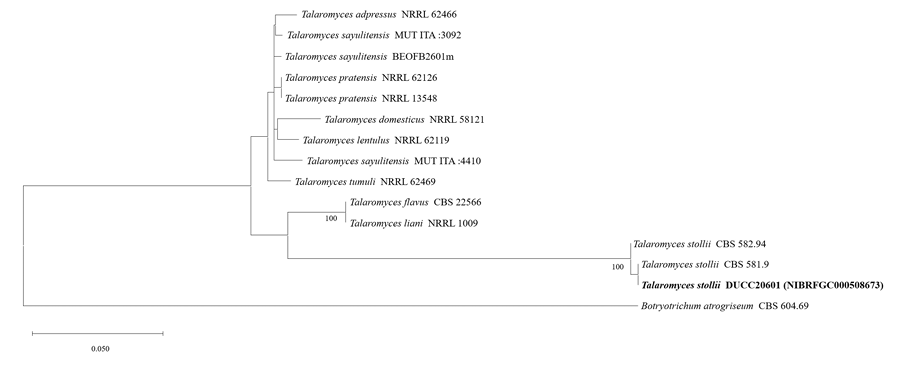
Talaromyces stollii DUCC20601 (NIBRFGC000508673) colonies grew to 32, 14, 27 and 33 mm on PDA, MEA, CYA, and OA, respectively, after 7 days. The colonies were green with a white border on PDA and CYA, white on MEA, and dark green with a white border on OA (Fig. 13). The DUCC20601 strain has 2-3 μm spherical conidia and 6-7 μm elongated cylindrical phialide. When observed under SEM, the conidia had a bumpy surface (Fig. 13). Based on the morphological characteristics and molecular phylogenetic analysis, DUCC20601 strain was identified as Talaromyces stollii, showing 99.87% β-tubulin gene sequence similarity to T. stollii (GenBank accession no. MH792959.1) (Fig. 14; Table 2). In 2012, T. stollii was isolated from a patient with acquired immunodeficiency syndrome in the Netherlands in 2012 and caused pineapple fruitlet core rot in 2020 [26-27].
Fig. 13
Colony morphology of Talaromyces stollii DUCC20601 (NIBRFGC000508673) strain grown on different media and their microstructures. A: Potato dextrose agar (PDA). B: Malt extract agar (MEA). C: Czapek yeast agar (CYA). D: Oatmeal agar (OA). E-G: Light microscopic images of conidia, phialides, metule and conidiophore. H: Scanning electron microscopy (SEM) image of conidia. Scale bar, E-G=10 µm, H=1 µm.
Microstructure observation: The microstructures of the isolated fungi were observed using an optical microscope (BX53; Olympus, Tokyo, Japan). The size of each microstructure was measured 50 times, with the range shown.
DNA extraction and phylogenetic analysis: Purely isolated fungi were cultured on PDA lined with sterilized cellophane for another week and approximately 50 mg of mycelia was scraped with a surgical blade and collected in a 2 mL tube. DNA was extracted from the collected mycelia using a Plant/Fungi DNA Isolation Kit (Norgen Biotek Corp., Thorold, Canada). Using the extracted DNA as a template, PCR primers in Table 1 were used to amplify the internal transcribed spacer (ITS) region (ITS1-5.8S-ITS2), the large subunit of nuclear ribosomal RNA (28S rDNA), and partial β-tubulin gene (BenA) sequences. PCR was performed under the conditions listed in Table 1 using a Bio-Rad T100 Thermal cycler. After confirming PCR product amplification by electrophoresis on a 0.8% agarose gel, the PCR amplicon was purified and sequenced by Macrogen Corp. (Seoul, Korea).
The determined nucleotide sequences were analyzed for homology using the National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov/) search engine GenBank BLASTN. Fungal species closely related to the taxa isolated in this study and belonging to the same or different genera, were included in the analysis. Reference sequences were obtained from GenBank and are listed in Tables 2-8. Individual sequence datasets of the ITS region, 28S rDNA, and the β-tubulin gene were aligned using the MUSCLE (multiple Sequence Comparison by Log- Expectation) alignment tool of MEGA XI [11] and improved manually where necessary using BioEdit [12]. Based on the aligned sequences, a maximum likelihood (ML) phylogenetic tree was constructed and the reliability of each node in the phylogenetic tree was evaluated using 1,000 bootstraps [13].
RESULTS AND DISCUSSION
The morphological characteristics of each species of unrecorded soil fungi isolated and identified in this study and the results of phylogenetic analysis based on nucleotide sequence analysis of ITS and 28S rDNA are presented below. The seven DUCC strains were deposited at the National Institute of Biological Resources of the Republic of Korea (https://www.nibr.go.kr/cmn/main/enMain.do) and received the NIBRFGC numbers. The analyzed nucleotide sequences were registered in GenBank of the NCBI database and the registration numbers of the seven DUCC strains are provided in Tables 2-8.
Acrocalymma walkeri Trakunyingcharoen et al. IMA Fungus 5(2):391-414 (2014) [MB#810840]
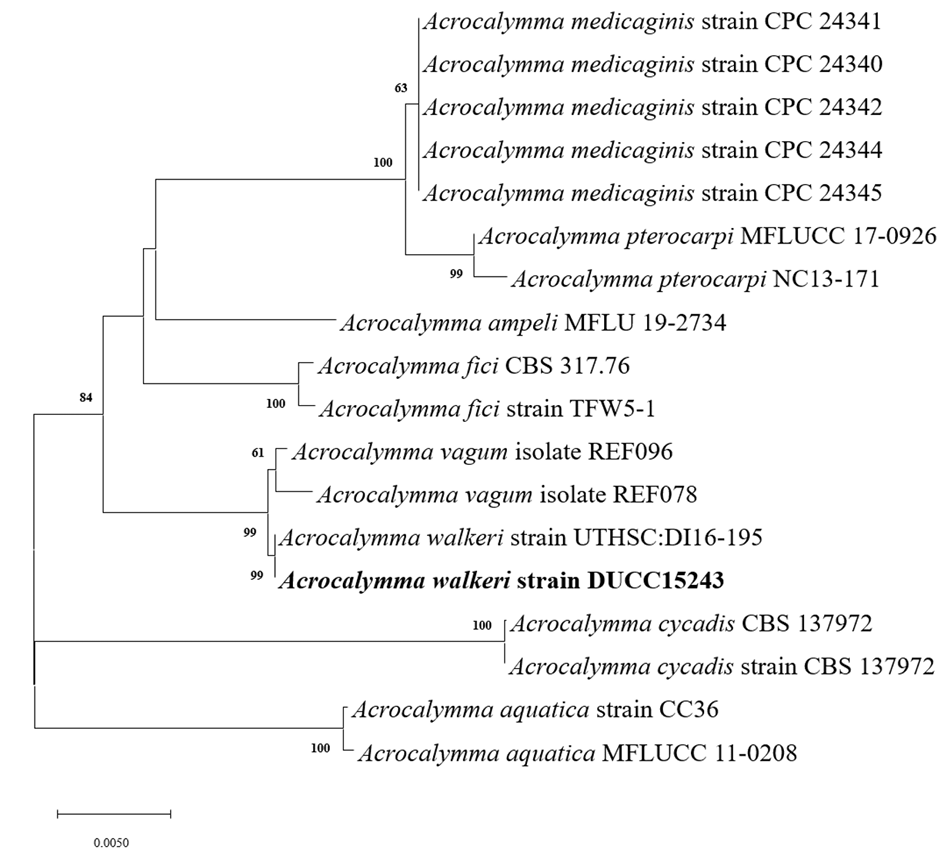
Macro morphological characteristics were observed after 7 d of incubation in the dark at 25℃. Acrocalymma walkeri DUCC15243 (NIBRFGC000509977) grew the best with a diameter of 38-40 mm on MEA and CYA and grew the slowest with a diameter of 26-31 mm on OA. The colonies were generally white, slightly fluffy, and circular and had few developed aerial hyphae. However, DUCC15243 showed a light pink colony color on OA. Microscopic morphological characteristics: Hyphae were hyaline, branched, septate, thin-walled, and 2-3 μm thick. Conidia were hyaline, with a smooth surface, without septum, olive colored, oval, filled with oily droplets, and 14.5-16.2×3.2-4.1 μm thick (Fig. 1). No sexual state was observed. A. walkeri was isolated from Medicago sativa in Queensland, Australia by Crous and Trakunyingcharoen in 2014 [14]. A. walkeri has been reported to have a similar morph whereas larger conidia (11-21×3.5-5 µm) compared to those of A. medicaginis. Molecular biological characteristics: The ITS region of the DUCC15243 strain showed 99.79% similarity with that of A. walkeri LT796832 and the 28S rDNA region showed 99.55% similarity with that of A. walkeri MH874057. According to the phylogenetic tree based on the combined ITS and 28S rDNA sequences shown in Table 2, DUCC15243 strain formed the same clade as A. walkeri strain UTHSC:DI16-195 (Fig. 2). Based on the morphological and molecular analysis, DUCC15243 strain was identified as Acrocalymma walkeri, an unrecorded species in Korea. A. walkeri causes root and crown rot in Medicago sativa [15]. A. vagum belonging to the same genus as A. walkeri produces dibenzo-α-pyrone derivatives with antibacterial and antifungal activity [16] and licorice, a medicinal plant, has been reported to tolerate drought stress better when its rhizosphere was treated with A. vagum [17]. Therefore, it would be interesting to study whether A. walkeri can produce substances with antibacterial and antifungal activities and interact with plants in the rhizosphere.
Table 2
Sequence information of species closely related to Acrocalymma walkeri used for constructing a phylogenetic tree based on the concatenated sequences of ITS and 28S rDNA region
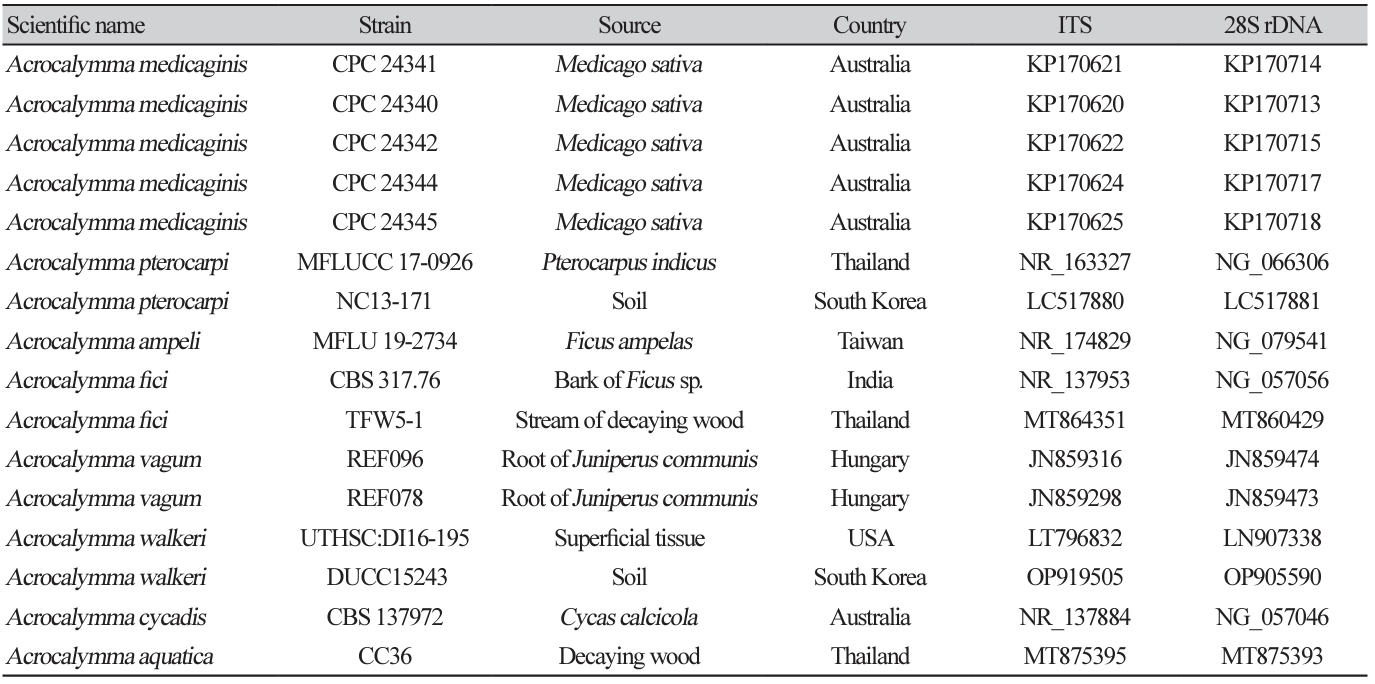
Fig. 2
Maximum likelihood phylogenetic tree based on the concatenated ITS region and 28S rDNA nucleotide sequences of Acrocalymma walkeri DUCC15243 (NIBRFGC000509977) strain. The number of nodes represents the reliability value through 1,000 bootstrap replicates. The fungal strain isolated in this study is indicated in bold.
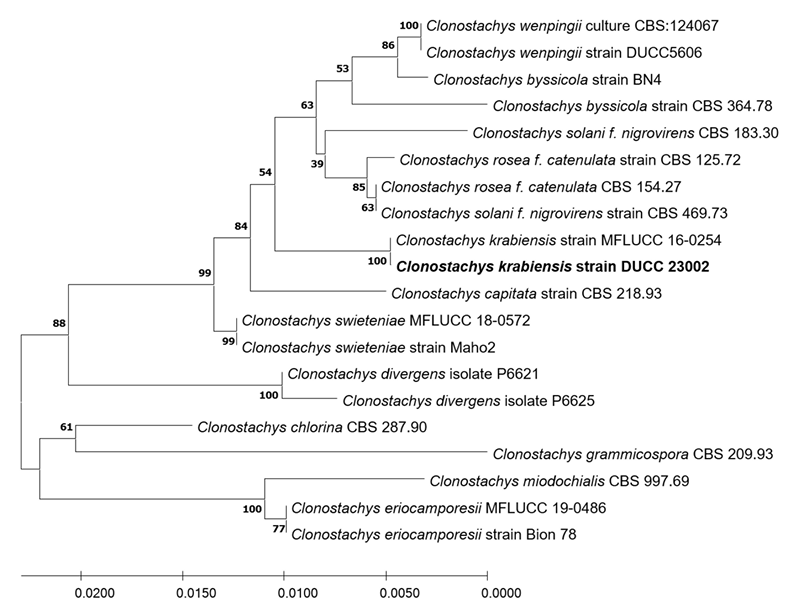
Clonostachys krabiensis Tibpromma et al. Fungal Diversity 93:1-160 (2018) [MB#554527]
Macro morphological characteristics were observed after 7 d of incubation in the dark at 25℃. Clonostachys krabiensis DUCC23002 (NIBRFGC000510016) were fluffy, circular, radial, and white and developed aerial hyphal morphologies on all media. The DUCC23002 strain grew best, with a diameter of 34 mm on PDA and showed the slowest growth on OA. Microscopic morphological characteristics: Hyphae were hyaline, branched, septate, thin-walled, and 1-3 μm thick. Conidiophores were hyaline and thin-walled; developed at the tip or middle of the hyphae with a size of 13.7-25.1×2.6-4.6 μm. Phialides were hyaline and thin-walled; 1-3 developed at the end of the conidiophore, with a size of 14.2-30.1× 2.3-4.5 μm. Conidia had a smooth surface, no septum, olive color, oval shape, and a size of 3.4-5.6×2.55.0 μm (Fig. 3). No sexual state was observed. C. krabiensis was isolated from Pandanus spp. in Thailand by Tibromma in 2018 [18]. C. krabiensis has been reported to have conidiophores that aggregate into sporodochia with hyaline setae around the margin. The conidiogenous cells were monophialidic, hyaline, subulate, and 10-13×1.5-2.5 μm in size. Conidia were slimy, solitary, aseptate, hyaline, cylindrical to oblong, smooth-walled, and 5-7×1-2 μm in size. Molecular biological characteristics: The ITS region of the DUCC23002 strain showed 98.49% similarity with that of C. krabiensis NR_168189 and the 28S rDNA showed 99.76% similarity with that of C. krabiensis NG_068835. According to the phylogenetic tree based on the combined ITS region and 28S rDNA sequences in Table 3, DUCC23002 strain formed the same clade as C. krabiensis strain MFLUCC 16-0254 (Fig. 4). Based on morphological and molecular analysis, DUCC23002 strain was identified as Clonostachys krabiensis, an unrecorded species in Korea. C. rhizophaga belonging to the genus Clonostachys is a plant pathogen that causes chickpea wilt [19]. As a fungal parasite, C. rosea has strong biological control abilities against numerous fungal plant pathogens, nematodes, and insects [20]. However, scarce information is available on C. krabiensis and additional research should be conducted to determine its value and role in nature.
Fig. 3
Colony morphology on different media (A–D) and light microscopic images (E-H) of Clonostachys krabiensis DUCC23002 (NIBRFGC000510016) strain grown at 25℃ for 7 d. A: PDA. B: MEA. C: CYA. D: OA. E-G: Conidiophore. H: Conidia. Scale bar=10 μm.
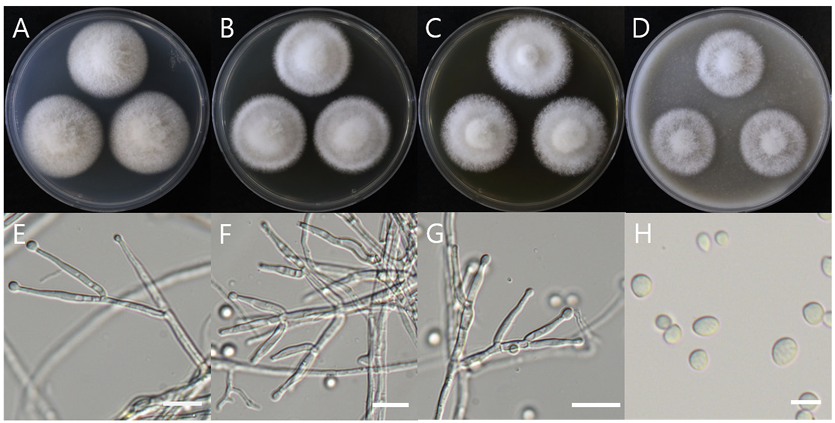
Table 3
Sequence information of species closely related to Clonostachys krabiensis used for constructing a phylogenetic tree based on the concatenated sequence of ITS and 28S rDNA region
Fig. 4
Maximum likelihood phylogenetic tree based on the concatenated ITS region and 28S rDNA nucleotide sequences of Clonostachys krabiensis DUCC23002 (NIBRFGC000510016) strain. The number of nodes represents the reliability value through 1,000 bootstrap replicates. The fungal strain isolated in this study is indicated in bold.